

By Nathifa Nasim, Neurobiology, Physiology, and Behavior ‘22
Author’s note: While in the Jin lab at the MIND Institute, I was introduced to the intersection between inflammation and neurodegeneration, specifically in the context of Alzheimer’s disease. My interest in this relationship has primarily been in manipulating inflammatory pathways to investigate the effects on the disease. However, I wanted to step back and understand how they connected, and compile a review on microglial activation as a bridge between the two.
Introduction:
Alzheimer’s, the most common form of dementia, is a neurodegenerative disease characterized by progressive loss of memory and cognitive function. It is still largely untreatable: the last drug approved by the FDA was released nearly two decades ago. Currently, there are only a few available treatments, all of which deal with alleviating symptoms rather than affecting any of the underlying pathology [1]. There are two primary hallmarks of Alzheimer’s disease: amyloid plaques and neurofibrillary tangles (NFTs). Amyloid plaques are formed from aggregations of small amyloid beta protein (Aβ); these amyloid beta are the product of cleavage of amyloid precursor proteins found in the membrane of neurons. Neurofibrillary tangles, on the other hand, form from tau protein, which stabilize neuronal microtubules and therefore allow for transport from the cell body to other parts of the neuron. Hyperphosphorylation of tau (a signaling mechanism) leads tao its detachment from the microtubule and aggregation into neurofibrillary tangles [2, 3].
Both amyloid plaques and NFTs are implicated in neurodegeneration and cognitive loss. Amyloid plaques are thought to be precursors that trigger a cascade culminating in neurodegeneration. On the other hand, the loss of support for microtubules in NFTs leads to impaired axonal transport, resulting in synaptic loss and neuronal dysfunction in an Alzheimer’s brain [2]. This review will explore an emerging aspect of Alzheimer’s research, microglial activation, as a means of mitigating both of these pathological characteristics of the disease thereby providing a potential avenue for approaching treatment.
The Role of Microglia:
In order to approach microglial activation, it is necessary to establish neuroinflammation’s role in neurodegeneration. Neuroinflammation refers to the central nervous system’s immune response, activated in response to trauma, pathogens, or the amyloid protein aggregations of Alzheimer’s, among others [2, 4]. It is a necessary immune response, but an overactive or continuous inflammatory response can be harmful, as evident in the body’s release of anti-inflammatory mediators alongside pro-inflammatory cytokines [2, 4]. Proinflammatory cytokines (proteins that are critical for immune signaling) such as IL-1β, IL-6, IL-18 and tumor necrosis factor (TNF), have various adverse effects on neuronal function including neuronal death, synaptic loss, and synaptic “pruning” or stripping [2, 5]. Therefore, unmitigated neuroinflammation can drive neurological disease, and is implicated in the pathology of all neurodegenerative diseases [4].
The main instruments of neuroinflammation are microglia: non-neuronal phagocytic cells that are the primary proponents of the brain’s immune response. Microglia recognize potential pathogens or irritants through receptors, and in response phagocytose and/or degrade the irritant while releasing cytokines, chemokines and interferons, immune signaling proteins [2]. There are two microglial activation states which dictate the inflammatory response: the “M1” or pro-inflammatory state associated with exacerbating neurodegeneration, and the “M2” or anti-inflammatory state [2, 4]. It must be noted that this binary is simplified, and currently under research. The overactivation of the inflammatory response can be linked to the M1 state of microglia. When inflammatory mediators such as IL-1β were released by microglia, they amplify the inflammation by activating more microglia, creating a positive feedback loop of neuroinflammation characteristic of a diseased state.
Based on their role in neuroinflammation, researchers have looked to microglia as key players in Alzhiemer’s pathology. Numerous research studies have indicated that microglial activation is increased in Alzheimer’s by observing increased expression of microglial receptors in the diseased brain [3, 6]. An example of this is a study that utilized [11C](R)-PK11195, a carbon labeled ligand specific to phagocytic cells. The ligand’s specificity to microglia was increased, allowing it to serve as an indicator for microglial activation. They found a significant increase of microglial activation in Alzheimer’s patients. Furthermore, the pattern of microglial activation physically mirrored the disease’s progress in the brain in terms of atrophy, among other indicators [5]. The research is supported by previous studies as well, all of which suggested that microglial activation is an early event in neurodegeneration, as it was present in mild/early cases of Alzheimer’s [3, 5]. The immune response appeared to escalate into causing more damage as the disease progressed [3].
Amyloid Plaques:
Decades-old research has confirmed the involvement of microglia in Alzheimer’s by demonstrating that microglia cluster around amyloid plaques. There is a progressive increase of activated microglia closer to dense plaque buildup, as well as a linear increase of activated microglia as overall plaque numbers increase [7]. As previously mentioned, amyloid precursor protein splicing leads to a beta amyloid protein; a derivative of the splicing, sAPP-α, has been shown to activate microglia. As microglia are activated by irritants, this falls in line with the general defense role of microglia. The sAPP-α protein, especially an Alzheimer’s-causing isoform which is more likely to aggregate, acts as a threat and thereby activates microglia. As an assumed consequence of the microglial activation, the same study verified that the presence of sAPP-α also increased inflammatory protein expression [8].
Tau Protein Involvement:
In addition to amyloid plaques, microglia have more recently been linked to the other hallmark of AD, neurofibrillary tau tangles. Similar to amyloid plaques, a linear pattern between NFT’s and activated microglia has also been shown [7]. Further supporting this connection, experimental depletion of microglia has led to decreased tau propagation [9]. Interestingly, although inflammatory mediators observed in one study were increased in patients with tau tangles and neurodegeneration, this was not the case in patients with only amyloid plaques. This highlights the importance of tau in microglial activation, as well as the difference in microglial relations between the two [3].
The interconnection between microglia and tau is proposed to be due to microglial phagocytosis of damaged neurons containing misfolded tau. The tau is secreted in exosomes, and these “seeds” of misformed tau protein are capable of inducing other tau to misfold and aggregate [6, 9, 10]. Although the exact mechanism of microglia engulfing tau is unclear, this theory fits with the overall degenerative pathology of Alzheimer’s in that microglia “prune” already damaged neurons and then engulf them. This increased tau phagocytosis and consequent release of misfolded tau increases overall NFTs, thereby further aggravating the disease state.
Tau, Amyloid and Microglia:
Microglia may also play a role in the pattern of tau accumulation and growth in the Alzheimer’s brain. Typically, as the disease progresses, NFTs “grow” in specific patterns or stages, culminating in the neocortex, the part of the brain devoted to higher cognitive functioning. This accumulation of plaques and NFTs in the neocortex is theorized to be the cause for dementia [6]. The propagation of tau, however, is still not fully understood—research is being conducted on whether microglial activation could be a cause. The current understanding of Alzheimer’s pathology via the amyloid cascade hypothesis suggests that amyloid plaques precede other aspects of Alzheimer’s pathology and neurodegeneration, and that tau tangles occur as a result of the “cascade” [2]. However, a recent study proposed that microglia could be the key player in this cascade. Microglia are theorized to act on Aβ, thereby increasing tau propagation, although they are not directly implicated in tau spread. Studies have shown a correlation between activated microglia with the development of cognitive impairment and dementia, supporting the theory that microglia are responsible for the tau propagation patterns seen as AD progresses [6]. This bridge between tau and amyloid via microglia-driven inflammation is further elucidated by another study. Researchers propose that microglial activation, intended to clear amyloid, additionally activates kinase pathways, specifically p38MAPK, directly/indirectly increasing tau phosphorylation, leading to neurofibrillary tangles [11].
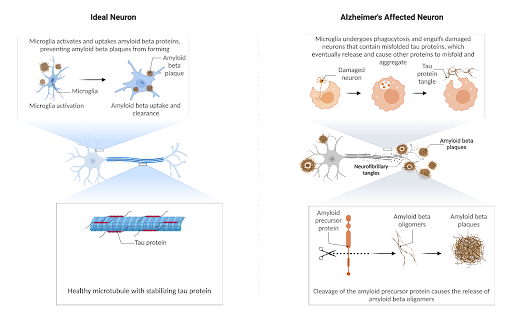
Figure 1. In the healthy neuron, tau stabilizes the neuron, but in the diseased state, the phagocytosis of misfolded tau culminates in the formation of more tau misfolding when it is released. For amyloid, specific cleavage sites result in oligomers prone to aggregation which ideally is phagocytosed by the microglia
Potential Influence on Treatment:
Based on the role of microglia in immune activation and its implication in Alzheimer’s pathology, inhibition of microglial activation could theoretically be neuroprotective against the disease, among other neurodegenerative diseases in which neuroinflammation plays a key role. Research published earlier this year expanded on this idea. Based on increased expression of a receptor in activated microglia found in Parkinson’s, a neurodegenerative disease similar to Alzheimer’s in that it is also marked by cognitive deficits, the researchers proposed utilizing the agonist NLY0. Not only did the administration of the agonist block microglial activation, it also reduced inflammatory mediators that in turn activate astrocytes, another glial cell, preventing the cycle of neuroinflammation to neurodegeneration. There were also reduced Aβ plaque numbers in an Alzheimer’s model, and perhaps as a result, improvements in cognition such as improved memory [1].
Conclusion:
Alzheimer’s disease is characterized by inflammation, through which microglia, as proponents of the brain’s immune response, are implicated in the development of the disease. The two main hallmarks of the disease — amyloid plaques and neurofibrillary tangles — are both associated with increased levels of activated microglia. However, in both cases, it is difficult to determine whether increased microglia are present as a result of neurodegeneration or whether they contribute to neurodegeneration. Nonetheless, emerging research places microglia as an important component of the amyloid cascade, by which Aβ and NFTs are connected. Neuroinflammation triggered by the need to clear amyloid plaques may lead to hyperactive kinase activity, hyperphosphorylating tau and leading to NFTs.
Given microglial involvement, further research is needed to investigate the potential of microglial inhibition in the treatment of Alzheimer’s, amongst other neurological diseases. However, the established interplay between microglia and Alzheimer’s pathology provides an important avenue in which to investigate related treatment options while illuminating the connection between inflammation and neurodegeneration.
References:
- Park JS, Kam TI, Lee S, et al. 2021. Blocking microglial activation of reactive astrocytes is neuroprotective in models of Alzheimer’s disease. acta neuropathol commun 9 (78). doi:10.1186/s40478-021-01180-z
- Leng F, Edison P. 2021. Neuroinflammation and microglial activation in Alzheimer disease: where do we go from here?. Nat Rev Neurol 17, 157–172. doi:10.1038/s41582-020-00435-y
- Nordengen K, Kirsebom BE, Henjum K, et al. 2019. Glial activation and inflammation along the Alzheimer’s disease continuum. J Neuroinflammation 16 (46). doi: 10.1186/s12974-019-1399-2
- Edison P, Donat CK and Sastre M. 2018. In vivo Imaging of Glial Activation in Alzheimer’s Disease. Front. Neurol. 9:625. doi:10.3389/fneur.2018.00625
- Cagnin A, Brooks DJ, Kennedy AM, Gunn RN, Myers R, Turkheimer FE, Jones T, Banati RB. 2001. In-vivo measurement of activated microglia in dementia, The Lancet, 358 (9280): 461-467. doi:10.1016/S0140-6736(01)05625-2.
- Pascoal TA, Benedet AL, Ashton NJ, et al. 2021. Microglial activation and tau propagate jointly across Braak stages. Nat Med 27, 1592–1599. doi:10.1038/s41591-021-01456-w
- Serrano-Pozo A, Mielke ML, Gómez-Isla T, Betensky RA, Growdon JH, Frosch MP, Hyman BT. 2011. Reactive glia not only associates with plaques but also parallels tangles in Alzheimer’s disease. The American journal of pathology, 179(3): 1373–1384. doi:10.1016/j.ajpath.2011.05.047
- Barger S, Harmon A. 1997. Microglial activation by Alzheimer amyloid precursor protein and modulation by apolipoprotein E. Nature 388: 878–88. doi:10.1038/42257
- Asai H, Ikezu S, Tsunoda S, et al. 2015. Depletion of microglia and inhibition of exosome synthesis halt tau propagation. Nat Neurosci 18: 1584–1593. doi:10.1038/nn.4132
- Hopp SC, Lin Y, Oakley D, Roe AD, DeVos SL, Hanlon D, Hyman BT. (2018). The role of microglia in processing and spreading of bioactive tau seeds in Alzheimer’s disease. Journal of neuroinflammation, 15(1): 269. doi:10.1186/s12974-018-1309-z
- Ghosh S, Wu MD, Shaftel SS, Kyrkanides S, LaFerla FM, Olschowka JA, O’Banion MK. 2013. Sustained interleukin-1β overexpression exacerbates tau pathology despite reduced amyloid burden in an Alzheimer’s mouse model. The Journal of neuroscience : the official journal of the Society for Neuroscience, 33(11): 5053–5064. doi:10.1523/JNEUROSCI.4361-12.2013