
By Mirabel Sprague Burleson, Biological Sciences ‘24
Cancer can contaminate nearly every tissue in the human body, arising from complex and diverse mutations that impact many genes. It’s incredibly widespread and lethal and is currently one of the most common causes of death in the United States, second only to heart disease by a small margin [1]. Despite the immense resources poured into cancer research, it remains a major issue in modern medicine. One barrier to the advancement of cancer treatments is finding a way to successfully treat cancer cells without harming the healthy cells surrounding them. Common treatments such as chemical and radiation therapy damage normally functioning cells, resulting in dangerous and debilitating side effects.
In order to develop a treatment that is capable of successfully isolating cancer cells, researchers must understand the hallmarks of cancer. Knowing the distinct characteristics of cancerous cells, such as abnormal division rate, increased mobility, and irregular organelles, allows researchers to develop treatments that attack these specific biological targets. Modern research focuses on determining unique bacterial presence in cancerous cells, as bacteria are an easy target for many treatments.
Recent studies on bacterial presence in cancer found metabolically active cancer-specific communities of bacteria in tumor tissues, which lead to their inclusion in updated cancer hallmarks [2]. In the wake of these findings, Narunsky-Haziza et al. (2022) conducted a study to determine if fungi could also be detected in tumor tissues [3]. Fungal presence in cancer cells could provide a new target for treatments.
The Narunsky-Haziza et al. study sources samples from four independent cohorts: The Weizmann Institute of Science (WIS), The Cancer Genome Analysis (TCGA), Johns Hopkins University, and the University of California at San Diego (UCSD) [3]. Narunsky-Haziza et al. took 17,401 tissue, blood, and plasma samples across 35 cancer types from these four cohorts. 104 samples made of a waxy substance called paraffin and 191 DNA-extraction negative controls were added to the WIS cohort samples to account for potential contamination by environmental fungi or fungal DNA introduced during handling and processing (other cohorts’ samples had adequate controls for fungal presence) [3,4,5]. These samples were then reexamined by Narunsky Haziza et al. for fungal presence with internal transcribed spacer 2 (ITS2) amplicon sequencing [3].
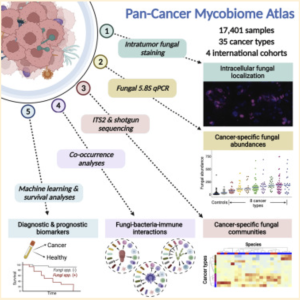
The ITS2 region of nuclear ribosomal DNA is considered one of the best DNA barcodes for sequencing because of its variability between even very closely related species and the ease of amplification [6]. ITS2 amplicon sequencing allows researchers to examine the ITS2 region and identify variations between samples. Narunsky-Haziza et al. use this method to cross-examine known fungal sequences and the sequences found in the samples to identify the different fungal nucleic acids present in the samples’ mycobiomes (fungal microbiome) [3].
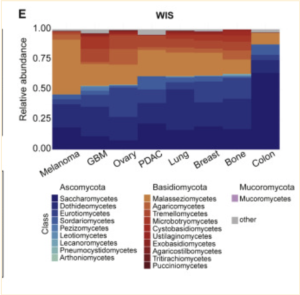
Using ITS2 amplicon sequencing, this study found that while tumor bacterial richness is much higher than fungal richness, there was a clear presence of fungi in the samples examined [3]. Fungi were detected in all 35 cancer types examined, although not all individual tumors were positive for fungal signals [3]. Most fungi were found to be within cancer and immune cells, similar to bacterial presence [3]. Interestingly, significant correlations were found between specific fungal presence and tumor types, immunotherapy, age, and smoking habits; however, whether this is correlated or casually associated is yet to be determined [3]. Also, an unexpected significant positive correlation between fungal and bacterial richness was found in bone, breast, brain, and lung samples, though not in any of the others [3].
This study does present several caveats. For one, differences in sample preparation, sequencing, bioinformatic pipelines, and reference databases exist between the four cohorts, which affect bacteriome analyses. Another potential issue is that although there was a large number of samples included, the stages of cancer across samples were different for all four of the cohorts, which created high variability in the data [4,5]. The WIS and TCGA cohorts also showed high variation in mycobiome richness, which Narunsky-Haziza et al. suspect is likely due to the negative controls introduced to the WIS cohort as well as potential split reads found in the TCGA cohort [3,4,5]. A split read is a sequence that partially matches the reference genome in at least two places, but has no continuous alignment to it. Split reads can indicate a difference in structure between the sample and the reference genome.

Additionally, while four different staining methods were used to find fungal presence and tumor-specific localization patterns, they proved to have differing sensitivities across cancer types. As all of the staining methods used can only detect certain subsets of the fungal kingdom, a relatively high false-negative rate can be expected. In contrast, although each cohort used negative controls, some false-positive results are inevitable [3].
Although this study successfully broadened the cancer microbiome landscape, these findings do not establish any causality in the presence of fungal nucleic acids. Narunsky-Haziza et al. hope that this first pan-cancer mycobiome atlas will serve as a key player in informing future cancer research to help characterize new information for cancer diagnostics and therapeutics [3]. While it remains unclear if fungal DNA plays a role in cancer development or severity, with further research, fungal presence could prove to be a helpful biomarker and potentially provide advancement in cancer treatments for the benefit of patients worldwide.