
By Barry Nguyen, Biochemistry and Molecular Biology ‘23 & Vincent Tran Neurobiology, Physiology, and Behavior ‘23
Authors Note: During my study abroad in South Korea, I was taken back by the number of people smoking cigarettes in the streets. As a country that valued health and beauty, I was surprised by the frequent sights of civilians smoking cigarettes. I then realized that not many people are aware of the cognitive effects cigarettes may induce. We both wrote this review in hopes of spreading awareness of the link that many are not cognizant of.
Introduction
Alzheimer’s Disease (AD) is an irreversible neurodegenerative disease that is characterized by neuronal loss, memory impairment, and cognitive dysfunction (Wallin et al. 2017). It is the main form of dementia in the aging population and it has been projected to quadruple in the coming century. AD pathogenesis is caused by a variety of factors–smoking cigarettes being a very common environmental factor in today’s society. Cigarettes contain thousands of organic compounds that have the capacity to induce adverse health effects (Wallin 2017). Epidemiological studies strongly show cigarette smoking as an important risk factor in AD (Yuen-Shan Ho et al. 2012). Smoking cigarettes not only doubles the risk for developing cognitive disorders, but it also accelerates the rate of cognitive decline. Approximately 2 billion people worldwide use tobacco products, mostly in the form of cigarettes (Durazzo et al. 2014) and given the current projection of AD, it is becoming increasingly paramount to further investigate the relationship between smoking cigarettes and cognitive decline. In this review, we delineate 3 strongly supported mechanisms of tobacco use that manifest and contribute to AD pathogenesis, as well as discuss the societal factors that underlie individual differences in severity of AD symptoms.
Smoking-Associated Neurological Pathologies
Increased Cerebral Oxidative Stress
Maintaining the biochemical integrity of the brain is essential for its normal functioning. Oxidative stress (OxS), in addition to its capacity to onset various vascular pathologies (Chavez et. al, 2007), may also impair cerebral biochemistry (Salim 2017). Oxidative stress (OxS) is a phenomenon that is caused by free radicals, or chemical entities with an unpaired electron. Free radicals are common by-products of metabolic processes, and among which are reactive oxygen species (ROS) that have undergone a one electron reduction (Chavez et. al, 2007). When an excess amount of (ROS), reactive nitrogen species (RNS), and other oxidizing agents are produced (Durazzo et. al 2014), and the antioxidant system is unable to keep up with such radical formation, oxidative stress occurs (Salim 2017). In general, the resulting physiological pathologies include modification of biomolecules, which results in defective cellular signaling and accumulation of malfunctioned proteins. With this cascade of negative events induced by OxS, the imbalance between production and detoxification of ROS can have serious consequences on many of our physiological systems, and in particular, the brain.
Cigarettes are composed of approximately 5,000 combustion products and contain high concentrations of free radical species (Durazzo et. al 2017). Smoking inhibits the synthesis of antioxidant species, thereby propagating free radical formation and the consequences of oxidative stress. The human brain, in particular, is vulnerable to OxS damage due to its high metabolism and relatively low antioxidant enzymes. With this said, smoking cigarettes may have profound consequences on the brain.
To detect oxidative stress in the brain and its differential manifestations among smokers and nonsmokers, scientists divided a group of 9 male rats into two groups: four in the control and five in the cigarette smoking group (Ho et. al 2012). The rats in one group were exposed to sham air, or “clean air” to serve as the control while the rats in the experimental group were exposed to cigarette smoke for one hour a day for 56 days. After 56 days, the rats were euthanized and brain tissue was retrieved.
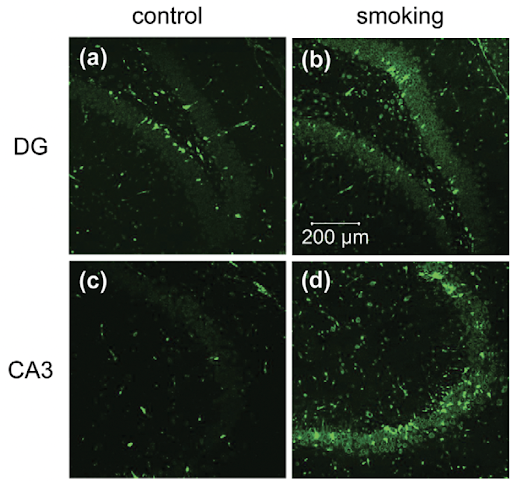
Figure 1. Anti-8-OHG antibody was used to visualize the levels of oxidative stress present in the hippocampus (Ho et. al 2012).
To detect levels of oxidative stress, researchers used anti-8-OHG, an antibody that stains biomarkers of oxidative stress and looked at regions in the brain responsible for memory. 8-hydroxy-2 deoxyguanosine (8 OHDG) and 8-hydroxyguanosine (8-OHG) are OxS biomarkers generated when guanine, a nucleotide present in DNA and RNA, is oxidized by reactive free radicals. In the IHC stainings, a higher fluorescence was observed among the experimental group as compared to the control, suggesting smoking cigarettes as a potential inducer in cerebral oxidative stress. Rats exposed to smoking show a significant immunoreactivity of 8-OHG in the dentate gyrus, which is a region in the brain responsible for memory (Fig. 1a and b), and CA3 (Fig. 1C and D) as compared to rats in the control group. The immunoreactivity of 8-OHG in the experimental group suggests that smoking does indeed induce oxidative stress by oxidizing the guanine nucleotide present in the DNA.
Decreased Expression of Synaptic Proteins
Synapsins are synaptic proteins that are essential for the normal functioning of the brain (Yuen-Shan Ho 2012). Synapsins belong to a family of phosphoproteins and are important for synapse development, neurotransmitter regulation, and nerve terminal formation (Mirza 2018). Current Alzheimer’s literature reveals a substantial involvement of malfunctioned synapsins in the development of AD. Namely, synaptic loss in the neocortex and limbic system, both regions important for higher order functions such as emotional responses, cognition, and spatial reasoning, may be responsible for the cognitive alterations in Alzheimer’s patients. Additionally, disturbances in synapsin homeostasis have revealed cognitive deficits and defective neurotransmitter transmission in Alzheimer’s patients (Lin et. al 2014).
In the same experiment investigating cigarettes’ capacity to induce neurological dysfunction, scientists observed a decreased expression of Synapsin 1, one of three isoforms of the protein (Ho et al. 2012). Using immunohistochemistry, the smoking group showed a reduction of fluorescent intensity, inferring decreased expression of Synapsin 1. These results further bolster the capacity of tobacco use in its contribution to AD pathogenesis due to the significance reduction in the Synapsin 1 protein as observed in the results. Furthermore, cigarettes’ ability to induce small scale pathological changes in the brain suggests its domino-like effect on cognitive function.

Figure 2. Immunohistochemical staining reveals significant reduction in Synapsin 1 protein between the control and smoking group (Ho et al. 2012).
Abnormal Phosphorylation of Tau Proteins
Tau pathology, a hallmark of Alzheimer Disease pathology, manifests due to the abnormal phosphorylation of Tau protein (Neddens 2018). This hyperphosphorylation of Tau results in Tau aggregation and are collectively known as neurofibrillary tangles (NFT), a histopathological marker for AD (Miao 2019). Oxidative stress in particular has a capacity to promote Tau pathology due to its fatty acid product which provides a direct link to mechanisms that induce NFT formation (Liu 2015). The mechanism in which OxS plays in the phosphorylation of Tau and subsequent aggregation is dependent on the type of oxidant and the specific amino acid sequences involved (Federica et. al 2019). For example, oxidation of cysteine residues have been observed to be involved in Tau aggregation, suggesting the phenomenon to be a disulfide bond mediated process.
Evidence linking oxidative stress and Tau hyperphosphorylation can be supported in an experiment utilizing Buthionine Sulfoximine treatment, which induces oxidative stress in M17 neuroblastoma cells (cancers of nerve cells) by inhibiting the synthesis of glutathione, a chemical important in the maintenance of the ROS equilibrium. Researchers were able to link HO-1, an oxidative stress biomarker, with an increase in hyperphosphorylation of PHF-1 sites. PHF sites, or paired helical filaments, are the structural constituents of neurofibrillary tangles, a pathological hallmark of AD. Taken together, smoking related OxS may serve as a fundamental mechanism in the pathogenesis of AD and indirectly influence AD pathogenesis by propagating the formation of NFT (Durazzo et. al 2014).
Possible Determinants of AD Differential Manifestations
As devastating as Alzheimer’s disease can be, the extent of its harm varies across a wide spectrum, and some people face greater damage or faster onset than others. Such variations in Alzheimer’s effects might be linked to not just biological but also environmental factors. As such, societal differences in the population can underpin the impact that various effects have on patients’ lifestyle and functioning with Alzheimer’s. This idea that Alzheimer’s effects are dependent on an individual basis is centered around an individual’s reserve against Alzheimer’s and other forms of dementia. Reserve against the effects of brain damage refers to the potential to alleviate dementia symptoms and progression and is further categorized into brain reserve and cognitive reserve.
Brain Reserve and Educational Attainment’s Connection to AD
Brain reserve describes how a larger amount of brain mass could offset the amount of damage that it would take for brain function to start being impaired. As patients afflicted by neurodegenerative diseases like Alzheimer’s could potentially lose more neurons and synapses before onset of clinical symptoms, those with larger brains could have better mitigation against symptoms of dementia (Bartrés-Faz et. al, 2011). With smoking linked to Alzheimer’s development, smokers could likely be diminishing the brain mass that would be buffering the rate at which neurological decay leads to impairment of memory and everyday functions.
Variations in individuals’ brain reserve could be associated with and predicted by individuals’ lifetime educational attainment. As such, a study using structural MRI analysis compared regional cortical thickness among a sample of individuals with different educational attainment. Those with more years of education were found to have larger regional cortical thickness, which was used to compare differences in brain size (Liu et al., 2012). This positive correlation between education level and cortical thickness demonstrates the positive impact of further education on development of more brain reserve.
Cognitive Reserve and Educational Attainment’s Connection to AD
Cognitive reserve is the concept that differences in learned cognitive processes can help the brain compensate for damage or dysfunction by relying on different functional approaches. As cognition for each individual relies on different recruitment patterns of neurons and synapses, individuals with more extensive neural networks would be more likely to compensate for the loss of neurons in a network vital for specific cognitive tasks (Bartrés-Faz et. al, 2011). Therefore, the same amount of brain damage to two individuals with similar brain sizes and physiologies could potentially result in differing effects in their functioning, due to differences in cognitive reserve.
Furthermore, differences in cognitive reserves can be attributed to lifetime educational or occupational levels, with epidemiological studies showing that individuals with less than 8 years of education have a significantly higher chance of dementia (Stern, 2012).
Access to educational opportunities has been unequally distributed across socioeconomic lines, with higher education’s high costs making it significantly more accessible to the middle and upper classes. Furthermore, there has also been a disparity in the racial distribution of Alzheimer’s in the U.S., with African Americans having the highest prevalence of AD followed by Hispanic Americans.
With smoking already presented as a risk factor for Alzheimer’s, its risk is compounded by the lessened reserve that is associated with education. Demographic studies demonstrated an inverse association between chances of smoking and educational attainment, with those who have less years of education being more prone to starting (Maralani, 2013). With those who smoke likely to have less education, such individuals would increase physiological risks of acquiring AD while also being more susceptible to developing AD at an earlier time.
Conclusion
Although smoking has been known to be the root perpetrator in a multitude of health risks and diseases, its effect on neurological health warrants increased scientific and public attention. As Alzheimer’s remains without a definite cure, current treatments revolve around managing symptoms and prevention of risk factors. With how smoking involves increased cerebral oxidative stress, decreased expression of synaptic proteins, and abnormal phosphorylation of Tau proteins, recent findings reiterate the necessity of avoiding smoking cigarettes to minimize further risks of developing AD. In the case that Alzheimer’s does develop in individuals, they can have a higher quality of life living with symptoms depending on their educational history, reiterating society’s need for better access to education.

Smoking cigarettes can produce 3 substantive effects that may contribute to the pathogenesis of AD: affecting synaptic proteins, increasing oxidative stress, and raising levels of hyperphosphorylated Tau protein.
References:
- Bartrés-Faz, D., & Arenaza-Urquijo, E. M. (2011). Structural and functional imaging correlates of cognitive and brain reserve hypotheses in healthy and pathological aging. Brain Topography, 24(3-4), 340–357.
- Durazzo, T., Mattsson, N., & Weiner, M. 2014 Smoking and Increased Alzheimer’s Disease Risk: A Review of Potential Mechanisms. Trauma Spectrum Disorder and Health Behavior 10:122-145
- Durazzo, T., Korecha, M., Trojanowski, J., Weiner, M., O’Hara, R., Ashford, J., & Shaw., L Active Cigarette Smoking in Cognitively-Normal Elders and Probable Alzheimer’s Disease Is Associated with Elevated Cerebrospinal Fluid Oxidative Stress Biomarkers. Journal of Alzheimer’s Disease 54: 99-107.
- Chavez, J., Cano, C., Souki, A., Bermudez, V., Medina, M., Ciszek, A., Amell, A., Vargas, M., Reyna, N., Toledo, A., Cano, ., Suarez, G., Contreras, F., Israili, Z., Hernandez-Hernandez, R., & Valasco. M. Effect of Cigarette Smoking on the Oxidant Antioxidant Balance in Health Subjects. American Journal of Therapeutics 14: 189-193.
- Ho, Y., Yang, X.,Yeung, S., Chiu, K., Lau, C., Tsang, A., Mak, J., & Chang, R. Cigarette Smoking Accelerated Brain Aging and Induced Pre-Alzheimer-Like Neuropathology in Rats. PLos ONE:7.
- Lin. L., Yang, S., Chu, J., Wang, L., Ning, L., Zhang, T., Jiang, Q., Tian, Q., & Wang, J. Region-Specific Expression of Tau, Amyloid-B Protein Precursor, and Synaptic Proteins at Physiological condition or Under Endoplasmic Reticulum Stress in Rats. Journal of Alzheimer’s Disease 41:1149-1163
- Liu, Y., Julkunen, V., Paajanen, T., Westman, E., Wahlund, L.-O., Aitken, A., Sobow, T., Mecocci, P., Tsolaki, M., Vellas, B., Muehlboeck, S., Spenger, C., Lovestone, S., Simmons, A., & Soininen, H. (2012). Education increases reserve against Alzheimer’s disease—evidence from structural MRI analysis. Neuroradiology, 54(9), 929–938.
- Maralani, V. (2013). Educational inequalities in smoking: The role of initiation versus quitting. Social Science & Medicine, 84, 129–137.
- Miao, J. Shi, R., Li, L., Chen, F., Zhou, Y., Tung, Y., Hu, W., Gong, C., Iqbal, K., & Fei, L. Pathological Tau From Alzheimer’s Brain Induces Site-Specific Hyperphosphorylation and SDS-and Reducing Agent-Resistant Aggregation of Tau in vivo. Frontiers in Aging Neuroscience 11: 34.
- Mirza, F. & S. Zahid. The Role of Synapsins in Neurological Disorders. Neuroscience Bulletin 34:349-358.
- Neddens, J., Temmel, M., Flunkert, S., Kerschbaumer, B., Hoeller, C., Loeffler, T., Niederkofler, V, Daum, G., Attems, J., & Paier, B. Phosphorylation of Different Tau Sites During Progression of Alzheimer’s Disease. Acta Neuropathol Commun 6.
- Salim, S. Oxidative Stress and the Central Nervous System. Journal of Pharmacology and Experimental Therapeutics 360:201205.
- Stern, Y. (2012). Cognitive Reserve in aging and Alzheimer’s disease. The Lancet Neurology, 11(11), 1006–1012.
- Wallin, C., Sholts, S., Osterlund, N., Luo, J., Jarvet, J., Roos, P., Llag, L., Graslund, A. & Warmlander, S. Alzheimer’s disease and cigarette smoke components: effects of nicotine, PAHs, and Cd(II), Cr(III), Pb(II), Pb(IV) ions on amyloid-β peptide aggregation. Scientific Reports 7.