

By Maya Mysore, Laura Ballou, Anna Rita Moukarzel, Alex Cherry, David Duronslet, Lisette Werba, Nathan Tran, Hannah Mosheim, Stephen Curry, Simon Coelho
Advisors: Kantharakorn Macharoen, Matthew McNulty, Andrew Yao, and Dr. McDonald, Dr. Nandi, and Dr. Facciotti
Author’s Note: My name is Maya Mysore, and I am a team lead on the BioInnovation Group’s Plant Bioprinter project. The BioInnovation Group is a student organization that creates research and leadership opportunities for undergraduates. The Bioprinter project is one of these opportunities.
I joined the BioInnovation group (BIG) in the winter quarter of 2019, as a freshman looking for ways to get involved on campus. I knew I liked research; I had been working in another lab. However, I was looking to explore different aspects of research. I heard about BIG through some friends in my major and went to an information session. There, I tried to join the more tech-based microfluidics project; however, my previous lab experience with cell culture convinced the lead for the Bioprinter project to get me involved in their work. I spent the next couple quarters investigating how to trap viruses in hydrogel. In Fall 2019, I was offered the role of lead. I was shocked, surprised, and a little out of my depth– after all, I had practically joined the project by accident! But I took on the role, excited about the leadership opportunity and the freedom. Now over a year into being project lead, I am planning to transition into the organization’s leadership. However, as a swan song to my time in charge, I wanted to compile all the hard work those involved with the project have accomplished. This paper is a celebration of the work of tens of student researchers over a period of several years. Hopefully, this paper will be the first of many for the Bioprinter project and the BioInnovation Group.
Abstract
As human space exploration expands to include potential settlement on the Moon and Mars, the ability to build shelter, manufacture food, produce medicine, and create other necessities in space will become increasingly important. Currently, the high cost and size constraints of sending payloads into space challenges us to think beyond the traditional manufacturing and agricultural tool-kit. Engineers have proposed that additive manufacturing, particularly 3D printing, is a solution to lower the payload costs and to enable the manufacturing of a variety of products in situ. This study focuses on 3D printing engineered biological cells for the production of biologics (e.g. pharmaceuticals that are living or derived from a biological source). We describe in-progress work to design, build, and test a small and affordable 3D bioprinter capable of printing 3D structured hydrogels that can carry living cells. We provide a general overview of the project, our progress in converting a low-cost and compact 3D printer from printing plastics to printing hydrogels, and preliminary work testing the compatibility of bioink formulations with genetically engineered rice cells that produce and secrete the enzyme butyrylcholinesterase.
Background
As humans continue to explore space and potentially settle in distant locations such as the Moon, Mars, and beyond, it will become increasingly necessary to build shelter, create food, and develop medicine while in space. However, the major costs (roughly $20,000/kg) and size constraints of sending payloads into space create challenges for such long-duration space travel beyond low Earth orbit [1-4]. Challenges include the manufacturing of food, shelter, and even medicine. 3D printing has been proposed as a cost-effective method for addressing some of these challenges, as it might allow the opportunity to ship only the printer to remote sites and to source the majority of the printing materials from the settlement location [5].
Biological systems may also play a large role in this approach. Microorganisms have been envisioned to help construct habitats through biocementation, a process that uses microorganisms to solidify inorganic matter into 3D structures [6-8]. Plants and microbes together are proposed as possible tools for the creation of sustainable ecosystems that recycle and detoxify waste and produce food [9-11]. A purported advantage of biological systems is that they can self-replicate, as each organism carries the full set of genetic instructions to create copies of itself. This means that biological systems could be delivered as light-weight “seeds”, i.e. self-replicating units that can be shipped in small and light quantities and grown to larger quantities upon permanent settlement at remote bases.
We and others envision that the 3D printing of engineered living systems (e.g bioprinting) may prove useful for the manufacturing of biologicals; this includes pharmaceuticals of or derived from a biological source [12]. In this context, the engineered living system serves as an on-demand expandable factory for the production of the biological while the 3D printer serves to produce custom-made culturing and purification hardware that can be produced in the geometries required for specific cells and production sizes. We were interested in exploring this concept and better understanding the challenges associated with the proposed process of drug production through bioprinting. In order to do this, we needed a bioprinter. Depending on their feature sets, commercial bioprinters can cost anywhere between $10,000 and $200,000, which was well outside our budget. Therefore, as a first step, we sought to design, build, and test a low-cost and compact bioprinter that we could later customize and use to explore novel design ideas.
FExisting modalities of bioprinting were considered and four main existing modalities of 3D bioprinting were considered: inkjet, pressure-assisted, laser-assisted, and stereolithography. For a detailed review on this subject, see Li et. al [13]. The major factors that were considered in the selection of a printer were types of usable bioinks, potential for good cell viability, cost, and complexity of the system (e.g. ease with which it can be modified). Inkjet-based bioprinting uses computer controls to drop small drops of bioink onto a surface. This type of printing maintains high short-tem cell viability and is widely available at low cost. However, it is limited in printing materials and creates high thermal and mechanical stress on cells which risks damage to cells and may affect long-term viability. Pressure-assisted bioprinters extrude bioink continuously onto a surface. While the extrusion process is slower and can lower cell viabilities immediately after printing (ranging from 40-80%, compared to 90% for inkjet printing), it allows use of a greater variety of materials and incorporates cells directly into the bioink. Laser-assisted bioprinters use a laser to irradiate a bioink such that the droplets adhere to the desired surface. This method of bioprinting is very precise and results in the highest cell viability; however, it is the most expensive, time-consuming, and has the highest risk of metal contamination. Finally, stereolithography printing uses illumination of a light-sensitive polymer to solidify 3D shapes. This method is fast, cost-effective, and has high final cell viabilities, but it is primarily limited by the need for a light-sensitive bioink, many of which are not biocompatible.
We chose to build a pressure-assisted bioprinter primarily due to practical factors: (a) the availability of low cost and compact fused deposition modeling (FDM) printers that could be used as chassis, theoretically enabling a “simple” swap of printing nozzles and pumps while taking advantage of the existing build platforms and 3D control systems; (b) the easy access to safe and low cost of compatible bioinks, and (c) the ability to incorporate cells directly into the bioink for prototyping.
This paper describes the progress of our project in developing a functional bioprinter. In addition, we describe the chemical assays used to evaluate engineered rice cell viability within hydrogels and these cells’ cell’s ability in gels to produce the pharmacologically-relevant enzyme Butrylcholinesterase (BChE), which is a complex human serine hydrolase enzyme that provides protection against organophosphorus poisoning from toxic agents such as sarin.
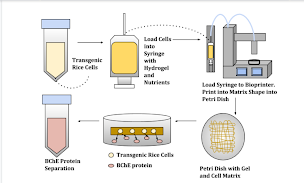
Figure 1. This diagram demonstrates the model methodology for seeding the cells into the hydrogel, printing out the cell-gel complex, and extracting the protein of interest from this system.
Methods
Printer selection, modification and testing
Selection of chassis
We sought to find a low-cost and compact FDM printer system that could be reasonably modified to extrude bioink rather than plastic filament. We ultimately selected the Monoprice MP Select Mini 3D Printer V2 because of its high availability, low cost ($250), and relative ease of modification. An accurate open source 3D computer-aided design (CAD) model (https://www.thingiverse.com/thing:2681912) of this printer was already available, making it easier to design new features for this specific unit.
Construction of an bioink extruder
To start converting the 3D plastic printer into a bioprinter, the printer’s original extrusion mechanism was replaced with a standard syringe/syringe-pump mechanism typical of bioprinters [14].
Incorporating the syringe-based bioink extruder required the design and construction of the entire extrusion system. An interchangeable mount was designed to hold the 10 mL syringe on the printer access, as seen in Figures 2b and 2c. In Figure 2b, the interchangeable mount design is shown with a trapezoidal connection piece, allowing the mount to swap between holding the 3D printer plastic extruder and the bioprinter syringe extruder system.

Figure 2. a) Inside of the 3D printer after all electrical components and panels were removed b) 3D printed interchangeable mount used to exchange the plastic extruder and the syringe extruder. c) The hydraulic extrusion system as connected to the bioprinter d) The hydraulic extrusion tubing system
The 10 mL syringe was connected to a hydraulic pumping system through a plastic tube. The hydraulic system is controlled using a Nema 17 Bipolar 40 mm Stepper Motor connected to an 8 mm threaded rod, forming a linear actuation mechanism. Connected to the rod is a 60 mL syringe plunger which is pushed through a 60 mL syringe. A liquid is placed in the 60 mL syringe and the bioink is placed in the 10 mL syringe also with a plunger sitting on top of the syringe. When the motor turns on, this liquid is pushed from the 60 mL syringe through the tubing and into the 10 mL syringe. This system pushes the plunger through the 10 mL syringe and extrudes the bioink onto the printing surface.
A T fitting made from 6 mm brass tubes was attached to the middle of the tubing system in order to remove air bubbles from the tube, as shown in Figure 2d.
Integration of hydraulic motors with chassis
To power the motor for the syringe extruder, the electrical components needed to be rebuilt. With this in mind, an Arduino Mega 2560 was connected with the HiLetGo RAMPS 1.4 control panel and the A4988 stepper motor driver boards using the wiring setup diagrammed in Figure 3.
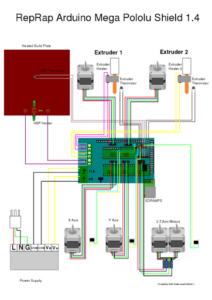
Figure 3. This diagram shows the wiring for the 3D printer using the Arduino.
The Z-axis switch was then repositioned and mounted to the printer chassis directly under the print head, as seen in Figure 2c.
Firmware
For the firmware, Marlin was selected because it is open sourced and easily modified with the Arduino IDE. After the firmware and electronics were set up, a G code file was needed to determine the print pattern. Cura was used to develop the file due to its compatibility with the Monoprice 3D printer. The Cura profile used with the bioprinter tests followed a cylindrical shape with a square-shaped infill grid. With this information established, the Cura profile was exported as G Code. In the printer design, an SD card is required to flash the firmware and upload the G code to the bioprinter. With the firmware and G code loaded onto the SD card, the bioprinter could be set up to run test prints with the bioink. The final cost spent to make the bioprinter came out to $375. Further information on the process of building the bioprinter can be found at https://www.instructables.com/Low-Cost-Bioprinter/.
Hydrogels
Hydrogels are porous water-based polymers that have many valuable uses, especially in fields such as drug delivery and tissue engineering. Here, we use hydrogels for their ability to selectively trap materials on a size basis, as this is what allows us to trap cells and release the protein of interest. Our hydrogel protocol was adapted from Seidel et al., 2017. Briefly, the hydrogel mixture contained agarose (0.2275% w/v), alginate (2.52% w/v), methyl cellulose (3% w/v), and sucrose (3% w/v). Agarose, alginate, and sucrose were mixed into deionized water at room temperature until dissolved. This mixture and the methyl cellulose powder were then autoclaved in separate containers for 20 minutes at 121 C. Upon completion of the autoclave cycle, methyl cellulose was mixed into the gel. The mixture was then left for 12 to 24 hours in a 2-8 C fridge to allow swelling to occur [15]. After this, the gel was ready to be seeded.
Seeding and Crosslinking the Gels
Transgenic rice cells were supplied by the McDonald-Nandi lab. The cells were genetically modified with the addition of a human BChE gene optimized for rice cell compatibility and cloned into the RAmy3D expression system for transformation into A. tumefaciens to allow insertion into rice cells [16]. This allowed the engineered cells to produce the pharmacologically-relevant BChE protein. The provided cell suspensions were mixed thoroughly via pipetting to obtain even distribution of cells. This suspension was then added directly to the hydrogel in a 50% volume split of cell suspension and gel and gently mixed to distribute cells evenly. To crosslink the gels and create solid structures for later use, a 0.1 M calcium chloride solution was prepared. The hydrogel was loaded into a syringe and deposited into weight boats containing enough CaCl2 solution to half-cover the extruded hydrogel. The hydrogel would then cure in the solution for at least 5 minutes or until the shape solidified. Upon completion of curing, the hydrogel could be removed and used for experiments.
Tetrazolium Chloride Viability Assay on Hydrogels
The TTC (2,3,5-triphenyltetrazolium chloride) assay is a method for testing cell viability. TTC is turned red from a colorless solution in the presence of metabolizing cells, allowing for quantification of cell viability. When used with defined standards and run on a spectrometer, it can be used to monitor cell survival over time.
Preparation of the TTC solution involved mixing 0.4% w/v TTC in 0.05 M sodium phosphate buffer, pH 7.5. Once the TTC solution was prepared, the TTC assay was performed.
5-6 mL of 0.05 M sodium phosphate buffer was added to a 15 mL Falcon tube with cured gel to submerge the cured gel entirely. The gel remained in the solution for 15 minutes. Then the Ellman buffer was removed from the tube and 500 μL of TTC were added to the tube with gel while mixing slightly. This tube was stored in a dark area for 24 hours.
If the gel was not cured, roughly 5 mL of gelled cells were first centrifuged in a 15 mL conical tube at 4500 g for 20 minutes. The supernatant was removed and 1 mL of Ellman buffer was added and mixed. The sample was centrifuged again at 4500 g for 15 minutes, the supernatant was removed, and 500 μL of TTC solution were added to the gel-cell mix. This sample was stored for 24 hours in a dark area.
After the 24 hours period ended, the sample-TTC mix was centrifuged at 4500 g for 15min. The supernatant was removed and the gel-cell mix was washed with 1 mL deionized water. The mixture was re-centrifuged at 4500 g for 10 minutes. The supernatant was removed again and 1 mL of 95% ethanol was added to the gel-cell mix. The sample was transferred to a microcentrifuge tube and placed in a 60C water bath for approximately 10 minutes. The sample is then centrifuged at 21.1 g for 15 minutes to recover the final supernatant. The supernatant was then run on a colorimeter or Tecan and the absorbance value was read at 485 nm. Beer’s law was then used to determine concentration from this value.
Seeded Cell-Ellman BChE Concentration Assay
The Ellman assay was used to measure BChE concentration for a sample at a given time point. This assay uses the kinetics of a color changing reaction to quantify the amount of BChE in solution. When in the presence of specific substrates, BChE turns a colorless solution yellow; the peak rate of this reaction can be determined and used to calculate BChE mass in a sample.
After cells were seeded into a hydrogel complex with a disc shape approximately 7 cm in diameter and 1 cm thick, the complex was suspended in 40 mL sucrose-free nutrient broth (NB-S).
The flask was then covered with a cloth filter and placed in the shaking incubator (37C, 5% CO2, 80 rpm). 50 μL media samples were collected from the flask daily over 14 days and the Ellman assay was run directly following collection of each of these samples.
The Ellman assay protocol was based on the Cerasoli lab protocol, which was adapted from Ellman et al., 1961 [17]. To perform the Ellman assay, a 20 mm stock solution of 5, 5’ – dithiobis-(2–nitrobenzoic acid) (DTNB) was prepared. A 75mM stock solution of S-Butyrylthiocholine (BTCh) iodide was also prepared.
Immediately prior to performing the Ellman assay, the Ellman substrate was prepared. 60 μL of DTNB and 30 μL of BTCh were added to the phosphate buffer in the falcon tube. The tube was temporarily stored in ice with light protection.
Then the Ellman assay was performed. In a 96-well plate, 50 μL of sample containing BChE was were diluted into 0.1 M phosphate buffer, pH 7.4, to ensure the generated? outputted slope readings (mOD/min) would fall in the range of 200-1000 when read for 3-5 minutes at 25 C. This dilution was done by estimating the approximate BChE concentration and estimating the mOD/min based on the expected value. 150 μL of Ellman substrate was added to each sample containing well. The optical density of the sample was immediately read at a wavelength of 405 nm for a total of 300 s (5 min) after the measurement was started.
After collecting data from the assay, Beer’s law was used to determine the concentration of product formed. From that value, we could estimate the mass of functional BChE in the total volume of the sample collected [18].
Results
TTC-Gel compatibility
To measure in-gel cell viability, we evaluated the use of the tetrazolium chloride (TTC) assay. This assay measures metabolic activity in live cells by reducing tetrazolium chloride to red formazan through the process of cell metabolism. Effectively, it provides an indication of how well the cells survive over time. Our team modified the assay for use in gels by including extra Ellman buffer and centrifugation steps to provide more opportunity for cells in the gel to be washed.
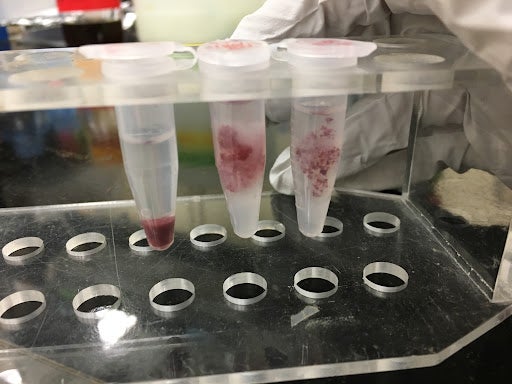
Figure 4. This figure shows the results of the TTC assay run on the transgenic rice cells in suspension. The leftmost tube is a positive control showing the TTC assay done on cell aggregates in suspension (i.e. without gel) that have been centrifuged into a pellet after the assay was performed. The middle and rightmost tubes are cells suspended in a hydrogel; the TTC assay was performed on this combination of cells in gels. In each tube, the cells have been stained red from the assay, indicating the presence of metabolic activity. These samples can go on to be washed and suspended in ethanol to obtain a viability data value.
To qualitatively assess how different factors like cell distribution and crosslinking might influence the results of the TTC assay, we performed additional variations of the assay. We first visually examined whether cell homogeneity was impacted by the gel. Then, we performed the TTC assay on E. coli cells alone as a positive control. After that, we tested the effects of non-crosslinked and crosslinked gel to ensure neither condition would prevent the use of the assay. E. coli was used for these tests due to our group’s ability to access it more regularly and grow it more easily than the genetically modified rice cells from Dr. McDonald’s lab. All of these tests together allowed us to determine that cell survival could indeed be measured within the gel, allowing us to monitor culture health over time. This will be critical in future use of the model, allowing us to determine ways to improve cell health and protein output by providing a metric for us to test against.
Homogeneous mixing of biological sample
To determine later TTC accuracy, the first key issue to address was homogeneity of cells in a hydrogel. This would determine whether sectioned off samples of cell-gel complexes would be representative of a whole sample. To ensure that the gel mixing protocol yielded a homogeneous suspension of the cells, we first tested our procedure by mixing E. coli expressing a transgenic green fluorescent protein (GFP) and imaged the suspension under UV light. We expect E. coli to distribute homogeneously in the gel similarly to the transgenic rice cells. This mixture was observed (Figure 5a) and confirmed by visual inspection of a homogeneous mix.
Test of TTC assay with bacterial suspension
To ensure that the TTC assay in later tests would be effective with E. coli, we first tested the TTC assay on an E. coli suspension as a positive control for later tests. We ran the modified TTC assay protocol described in Methods, and observed a color change in the solution. The resultant red solution (Figure 5b) matches the literature expectations for the output of this assay on living cells and indicates the assay is effective for E. coli.
Test of TTC assay with bacteria seeded in hydrogel
After confirming the TTC assay was effective with E. coli, it became important to determine how the presence of gel would affect the assay. We suspended the E. coli cells in the hydrogel and ran the modified TTC assay. The results seen in Figure 4c show the suspension turning red, which visually indicates the presence of cell metabolic activity and the effectiveness of the TTC assay.
Test of TTC assay with bacteria seeded in a crosslinked hydrogel
Upon determining the gel did not qualitatively affect the output of the TTC assay, it became necessary to determine whether crosslinking the gel had any effect on the effectiveness of the TTC assay. We reran the same experiment as the non-crosslinking hydrogel experiment, with the only change being the crosslinking process and the different first wash step. We found that the result of the TTC assay appears to be unaffected by the presence of the crosslinked out layer, as the solution turns red in the same way it does for the positive control and the non-crosslinked gel (Figure 5d).
These experiments allowed us to qualitatively determine whether the TTC assay could be an effective measure of cell viability. They also demonstrated that the introduction of a crosslinked hydrogel will not have visible impacts on measuring cell viability.
Figure 5. Qualitative TTC assays were run on E. coli with the pMax plasmid to test homogeneity within the gel and the effectiveness of the TTC assay in different hydrogel conditions. 5a shows the bacteria mixed homogeneously within the hydrogel, which is visible in the fluorescence that is present homogeneously through the sample. 5b shows the ethanol suspension output for a TTC assay run on a pMax E. coli culture, providing a control for later experiments and showing that the TTC assay is effective for E. coli. The left image is the control and the right image is the test condition. The control is run in the same conditions as the test, except the cells are placed in a 60C water bath for ten minutes prior to adding TTC in order to kill them. 5c shows the output prior to ethanol suspension for a TTC assay on E. coli pMax cells that were suspended in an uncured hydrogel. The left tube is the control and the right tube is the experimental condition. The red color visible in the right tube shows that the presence of the hydrogel does not prevent use of the TTC assay. 5d shows the ethanol suspension output for a TTC assay run on E. coli pMax cells that had been suspended in a cured hydrogel. The left image is the control and the right image is the experimental condition. The red color of the suspension indicates the TTC assay remained effective even with the addition of the crosslinked outer layer of the gel. Throughout this figure, variation in intensity of the redness of the samples is related to variations in time spent in suspension of the TTC solution, with redder samples correlating to longer time.
Initial Attempts at Measuring BChE Production
Our second major goal was to determine whether BChE could be collected from our model system (as seen in figure 1). This would allow us to determine if our model system was an effective way to collect our protein of interest for future space travel applications, as well as confirm that our test for BChE quantity would be effective in this system. To test this, our team ran the seeded cell-Ellman assay as described in methods to assess the amount of BChE that was escaping into the media. We first prepared a hydrogel, mixed the transgenic rice cells in, and cured it into a disc shape roughly 7 cm in diameter and 1 cm in height. We then suspended this cured cell-gel complex in NB-S media to stimulate BChE production, and we kept this mix in a spinning incubator to ensure aeration and adequate diffusion of materials in and out of the gel. Media samples were collected over the course of 14 days and were run with the Ellman assay for BChE detection on a spectrometer. The Ellman assay uses the enzyme kinetics of a color-changing reaction between BChE and a substrate to quantify the amount of BChE present in a sample at a given time point.
It is important to note that this test was intended as a trial run of the system in order to ensure that the assay works and that useful data is being collected. In addition, we sought to assess if BChE could escape from the gel at all. Therefore, no negative control was run and only one run of data was collected (shown in the figure below.) As a result, we cannot conclusively state anything about the data. However, the data does show a trend worth noting for future experimentation. The This is that active BChE concentration in the media increased for the first roughly 100 hours, after which the values dropped off. At the time point marked in figure 5, 96 hours, we see the maximal BChE present. If the unusually low value seen at the roughly 120 hour time point is considered erroneous (which we suspect), the data suggests increased production of BChE over the first 4 days of culture followed by a slow decay thereafter with production ending at around day 8. This provides an early quantitative estimate of the time-dependence of BChE production in this model system. This experiment is a first attempt and will be repeated with various parameter variations in the future.
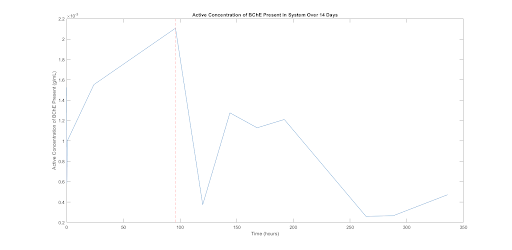
Figure 6. This plot shows the approximate active concentration of BChE released into the media for various time points over 14 days. Each sample was a 50 μL amount of media pulled from the small scale system model. This figure shows a burst in production of active BChE until the 96 hour time point (denoted with a dashed red line), after which the values drop off. The data point at t=120 hours is most likely an outlier resulting from this data being for one set of samples from one test condition.
Preliminary Bioprinter Testing
The process of building and testing the bioprinter was done in parallel with the TTC and Ellman assay testing. Detailed bioprinter testing has not been performed; however, initial testing of the printer showed its ability to print hydrogel into pre-programmed patterns. The grid pattern seen in Figure 7 was printed into a petri dish containing CaCl2 curing solution. The print shows excess hydrogel accumulation near the edges where the printhead briefly paused and reversed direction. In the center of the print, the lines in the grid averaged 1.25mm +/- 0.4 in width. Further testing and refinement is currently in process.
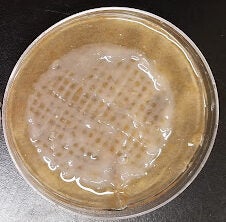
Figure 7. This shows a test print from our modified 3D printer using the hydrogel described in the methods section and cured in standard CaCl2 curing solution. This structure is described as a lattice shape and will be the primary pattern for future prints.
Discussion
In these experiments we determined that the TTC assay was effective in hydrogels, the Ellman assay showed the ability of protein to be detected from solution, and the bioprinter was able to create the desired lattice shape for later use.
Printer Performance
Our experiments to date have demonstrated our ability to convert a low-cost and compact FDM printer into a preliminarily functional bioprinter. The conversion of the original chassis required the modification of the printhead support, the development of a syringe-based hydraulic pump, and the modification of electronic and software control systems. Preliminary prints indicate that the printer can successfully deposit a programmed pattern with feature sizes in the range of 1.5mm. Existing conventional commercial bioprinters can achieve resolutions of 100-200µm, (some even claim filament diameters as low as 3µm), suggesting that we have room to improve the resolution of our system [19]. In addition to improving the resolution of the prints, we want to explore alternate methods for delivering the CaCl2 curing solution during alginate filament deposition to minimize user interaction and allow complete processing inside a biosafety cabinet; this should allow us to increase sterility during printing and print quality.
Cell Viability
Since it is known that pressure-assisted printing may negatively impact cell viability during printing, a key concern was the resulting cell viability of the system. As a result, our general goal for this phase of the project was to test whether a pressure-assisted bioprinter system could maintain cell viability after extrusion. We adapted the TTC assay for this purpose and tested our protocol to determine the effect of bioink and extrusion on cell viability under conditions mimicking those experienced during bioprinting.
Generally, the TTC assay demonstrated the ability of the assay to cellular viability in the crosslinked hydrogel, despite the unknown nature of how crosslinking affects pore size. Despite this success, the TTC assay remains largely qualitative as it is challenging to get quantitative measurements of cell viability when cells are embedded in a gel. This is further complicated by factors like the heterogeneous distribution of cells (or cellular aggregates) in the gel (see figure 4, rightmost sample). If homogeneity is not maintained, we need to design assays that take into account heterogeneous distribution of cells in the gel. In future experiments, we seek to determine whether samples from a large complex of cells in a hydrogel will provide a representative sample.
In later experiments, additional key variables that may potentially affect viability will be tested. These variables include media composition, culture duration, environmental conditions such as temperature, gel architecture, and the additional variables associated with the printer extrusion process (e.g. pressure, needle pore diameter, etc.). Determining how these specific factors affect viability will allow us to modify the printer design to minimize the drop in cell viability upon extrusion.
Protein production
Having confirmed the effectiveness of the TTC assay in the hydrogel, we moved forward to analyzing BChE production and its diffusion into the media. The assay we adopted allowed us to develop a standard method for data collection that can be used to analyze how various factors impact the cells’ ability to produce BChE. Figure 6, for example, shows that we can measure BChE production and diffusion out of the gel, and that under our preliminary experimental conditions, production peaks at 96 hours and then falls over the next 150 hours. While encouraging, this experiment needs to be repeated with many more samples and replicates to obtain a more reliable assessment of measurement error associated with the assay. Despite needing to replicate the experiment, we are confident that this preliminary experiment answered the core question of whether such a large protein – 85 kDa monomers and 4 units in quaternary form, with a total size of 574 monomers [20] – can effectively diffuse out of the hydrogel and avoid denaturation long enough to be collected and purified.
In addition to replication, future experiments should be explored to further improve protein escape from the hydrogel. These tests could increase the mixing speed to use centrifugal force to free proteins from the gel, increase pore size to create more physical space for protein escape, or print the 3-dimensional lattice structure to increase surface area and allow greater escape. Other relevant variables whose impact on BChE production should be tested include media composition and media changing schedules, culture duration, environmental conditions, gel architecture, and growth temperature. In our initial experiments, plant cells were grown in a shaking incubator at 37C to mimic the environment of protein production in mammalian hosts. However, this growth condition may have stressed the plant cells for which growth at 27C is more typical [16, 21]. This may explain the trend shown in figure 6, where die-off occurs after 96 hours.
Finally, in our current studies, the presence of sucrose in gel formulation (which inhibits BChE production) may have adversely impacted the amount of protein produced. While we expected that overlaying a relatively large volume of sucrose-free media would effectively dilute the sucrose to low levels, the presence of sucrose in the initial formulation could have nevertheless impacted the cells’ initial states and therefore protein production. A followup experiment that more stringently controls for the presence of sucrose in the gel than in the studies described above seems warranted.
Conclusion
In this work, we successfully modified an off-the-shelf pressure-assisted 3D printer into a working bioprinter. In addition, we established that BChE producing rice cells are biocompatible with the different bioink gel formulations and that our assays for testing cell viability and protein production are effective when analyzing the cells within the gel. Having shown that we can print gel, assess cell survival, produce BChE, and quantify its abundance, we next seek to optimize both printer function and the measurement assays for cell viability and protein concentration in ways that provide more quantitative data and more refined control over printed structures. Eventually, we expect that such advances will allow us to optimize protein production itself and ultimately develop a bioprinter suitable for protein production during space travel or in other remote locations.
Acknowledgements
Thank you to the Molecular Prototyping and BioInnovation Lab for the lab space, the BioInnovation Group for the administrative, scientific, safety, and monetary support, the McDonald-Nandi lab for materials and mentorship, and all past, present and future members of the Bioprinter team for contributing to these experiments.
References:
- Joshi SC, Sheikh AA. 2015. 3D printing in aerospace and its long-term sustainability. Virtual Phy Prototy [Internet]. 10(4):175–185. doi.org/10.1080/17452759.2015.1111519
- Hodkinson PD, Anderton RA., Posselt BN, Fong KJ. 2017. An overview of space medicine. Br J Anaesth [Internet]. 119(suppl_1):i143–i153. doi.org/10.1093/bja/aex336
- Aglietti, GS. 2020. Current Challenges and Opportunities for Space Technologies. Front Space Tech [Internet]. 1:1. doi.org/10.3389/frspt.2020.00001
- Coopersmith J. 2011. The cost of reaching orbit: Ground-based launch systems. Space Policy [Internet]. 27(2):77–80. doi.org/10.1016/j.spacepol.2011.03.001
- Ghidini T. 2018. Regenerative medicine and 3D bioprinting for human space exploration and planet colonisation. J Thorac Dis [Internet]. 10(Suppl 20):S2363–S2375. doi.org/10.21037/jtd.2018.03.19
- Gleaton J, Lai Z, Xiao R, Chen Q, Zheng Y. 2019. Microalga-induced biocementation of martian regolith simulant: Effects of biogrouting methods and calcium sources. Constr Build Mater [Internet]. 229:116885. doi.org/10.1016/j.conbuildmat.2019.116885
- Pilehvar S, Arnho M, Pamies R, Valentini L, Kjøniksen AL. 2020. Utilization of urea as an accessible superplasticizer on the moon for lunar geopolymer mixtures. J Clean Prod [Internet]. 247:119177. doi.org/10.1016/j.jclepro.2019.119177
- Kumar A, Dikshit R, Gupta N, Jain A, Dey A, Nandi A., … Rajendra A. 2020. Bacterial Growth Induced Biocementation Technology, ‘Space-Brick’ – A Proposal for Experiment at Microgravity and Planetary Environments. BioRxiv [Internet]. 2020.01.22.914853. doi.org/10.1101/2020.01.22.914853
- Lopez JV, Peixoto RS, Rosado AS. 2019. Inevitable future: space colonization beyond Earth with microbes first. FEMS Microbiol Ecology [Internet]. 95(10). doi.org/10.1093/femsec/fiz127
- Bornemann G, Waßer K, Tonat T, Moeller R, Bohmeier M, Hauslage J. 2015. Natural microbial populations in a water-based biowaste management system for space life support. Life Sci Space Res [Internet]. 7:39–52. doi.org/10.1016/j.lssr.2015.09.002
- Bokulich NA, Lewis ZT., Boundy-Mills K, Mills DA. 2016. A new perspective on microbial landscapes within food production. Curr Opin Biotechnol [Internet]. 37:182–189. doi.org/10.1016/j.copbio.2015.12.008
- Varma A, Gemeda HB, McNulty MJ, McDonald KA, Nandi S, Knipe JM. 2021. Bioprinting transgenic plant cells for production of a recombinant biodefense agent. BioRxiv [Internet]. 2021.02.01.429263. doi.org/10.1101/2021.02.01.429263
- Li J, Chen M, Fan X, Zhou H. 2016. Recent advances in bioprinting techniques: approaches, applications and future prospects. J Transl Med [Internet]. 14:271. doi.org/10.1186/s12967-016-1028-0
- Pusch K, Hinton TJ, Feinberg AW. 2018. Large volume syringe pump extruder for desktop 3D printers. HardwareX [Internet]. 3:49–61. doi.org/10.1016/j.ohx.2018.02.001
- Seidel J, Ahlfeld T, Adolph M, Kümmritz S, Steingroewer J Krujatz F, … Lode A. 2017. Green bioprinting: extrusion-based fabrication of plant cell-laden biopolymer hydrogel scaffolds. Biofabrication [Internet]. 9(4):045011. doi.org/10.1088/1758-5090/aa8854
- Corbin JM, Hashimoto BI, Karuppanan K, Kyser ZR, Wu L, Roberts BA, … Nandi S. 2016. Semicontinuous Bioreactor Production of Recombinant Butyrylcholinesterase in Transgenic Rice Cell Suspension Cultures. Front Plant Sci [Internet]. 7:412. doi.org/10.3389/fpls.2016.00412
- Ellman GL, Courtney KD, Andres VJ, Feather-Stone RM. 1961. A new and rapid colorimetric determination of acetylcholinesterase activity. Biochem Pharmacol [Internet]. 7:88–95. doi.org/10.1016/0006-2952(61)90145-9
- Alkanaimsh S, Karuppanan K, Guerrero A, Tu AM, Hashimoto B, Hwang MS, … McDonald KA. 2016. Transient Expression of Tetrameric Recombinant Human Butyrylcholinesterase in Nicotiana benthamiana. Front Plant Sci [Internet]. 7:743. doi.org/10.3389/fpls.2016.00743
- Gu Z, Fu K, Lin H, He Y. 2020. Development of 3D bioprinting: From printing methods to biomedical applications. Asian J Pharm Sci [Internet]. 15(5):529–557. doi.org/10.1016/j.ajps.2019.11.003
- Ngamelue MN, Homma K, Lockridge O, Asojo OA. 2007. Crystallization and X-ray structure of full-length recombinant human butyrylcholinesterase. Acta Crystallogr [Internet]. 63(9):723-727. doi.org/10.1107/S1744309107037335
- Macharoen K, McDonald KA, Nandi S. 2020. Simplified bioreactor processes for recombinant butyrylcholinesterase production in transgenic rice cell suspension cultures. Biochem Eng J [Internet]. 163:107751. doi.org/10.1016/j.bej.2020.107751