

By Surya Vishnubhatt, Biomedical Engineering, ’22
Author’s Note: The devastating COVID-19 pandemic, having resulted in the death of millions of people worldwide, has spurred innovation in countless sectors of academia, namely in the field of bioinformatics and computational biology. By using computer science techniques, researchers have been able to rapidly identify treatments and further analyze the SARS-CoV-2 virus; the following review synthesizes computational advancements in COVID-19 research through immunoinformatics, docking servers, machine learning, and microRNA analysis. This review also incorporates current computational approaches in the treatment and analysis of COVID-19 viral variants. The use of bioinformatics and computational biology, in pursuit of analyzing and treating all forms of COVID-19, has yielded fast and effective therapeutic treatments in conjunction with crucial analytical findings. With much of the United States now opening up, and the virus likely to become a global endemic, the need for fast, computational analysis of the disease, regarding its progression and spread, is invaluable in ensuring public safety.
Introduction
The COVID-19 pandemic is a global public health emergency, with the fast spreading virus having engulfed the world within a few months. The respiratory disease, as of February 2022, has resulted in the death of 5.9 million people worldwide [1]. The viral pathogen behind COVID-19 is SARS-CoV-2 (severe acute respiratory syndrome coronavirus 2). This relentless virus has a mutation rate of 9.8 x 10-4 substitutions per site per year, which refers to the replacement of a specific DNA base pair with another by means of nucleotide substitution. Given the scale of the genome (the human genome is 3.2 billion base pairs long), the mutation rate of SARS-CoV-2 allows for substantial changes to the virus’ spike protein, allowing it to evade its host’s defenses and causing new viral variants to crop up around the world [2]. As the prevalence of these variants increases worldwide, the importance of effective computational analysis of COVID-19 protein and antibody dynamics cannot be overstated.
Bioinformatics and computational biology are highly similar, interdisciplinary areas of study that use the core tenets of computer science to analyze biological data. In general, bioinformatics and computational biology are crucial in understanding and analyzing protein dynamics, primarily in regards to sequence, structure, and evolution based analysis (which tracks changes in protein composition over time) [3].
A variety of computational approaches have been and are being investigated in the hopes of better understanding how COVID-19 operates in order to develop effective therapeutic treatments. One such approach concerns the field of immunoinformatics, a subset of bioinformatics and computational biology, which uses protein structures and genome sequences to develop vaccines [4]. Other areas of interest involve the use of docking servers (which predict and model protein-ligand binding interactions) and machine learning to identify and develop COVID-19 therapeutics [5]. Further research is also being conducted in analyzing micro RNA to better understand and exploit the cellular dynamics of COVID-19 [6].
This review will investigate established computational approaches and will also explore novel research regarding COVID-19 in the hopes of stimulating further research in COVID-19 variant analysis.
A Brief History of COVID-19
The SARS-CoV-2 virus emerged in December of 2019, first reported in Wuhan, China. The unique virology of SARS-CoV-2 allowed for its rapid progression and spread. The virus itself is covered with spike proteins on its surface; each spike protein consists of three monomeric units which bind to human ACE2 receptor cells [7]. ACE2 receptors are present on the surface of human muscle cells, primarily in the lungs, and act to mediate vascular constriction and inflammation. During COVID-19 pathogenesis, the SARS-CoV-2 spike protein can bind to the human ACE2 receptor, invade the cell, and proliferate, leading to lung damage. The spike protein has an incredibly high affinity for the ACE2 receptor due to contact interactions which occur at the interface between the receptor binding domain of the spike protein and the ACE2 receptor, contributing to the widespread nature of the disease [8].
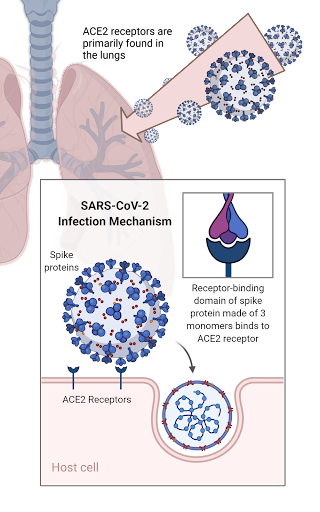
Figure 1: The SARS-CoV-2 infection mechanism.
Currently, in the United States, vaccines have been created for the original viral strain, namely the double dose Pfizer and Moderna (mRNA based) vaccines and the single dose Johnson and Johnson (adenovirus based) vaccine [9]. These vaccines stimulate the host to synthesize a non-pathogenic version of the spike protein, which triggers an immune response and is then targeted by host-specific antibodies (generated in response to the host-mediated spike protein), rendering immunity to the major COVID-19 strain. However, with the rise of new variants, most notably, the omicron B.1.1.529 strain, the effectiveness of mRNA (e.g. Pfizer and Moderna) vaccines are diminishing from 95% efficacy to 35% efficacy, with booster shots required to increase efficacy to 75% [10, 11]. Similarly, the Johnson & Johnson adenovirus based vaccine declined from 94% to 85% efficacy in adenovirus based vaccine therapies in individuals who received booster shots [12, 13].
Applications of Bioinformatics and Computational Biology in COVID-19 Research
The field of bioinformatics and computational biology deals in the collection and analysis of biological data, namely genomic and proteomic data, in the hopes of better understanding disease pathogenesis [3].
3.1 Immunoinformatics and COVID-19 Vaccine Development
The field of immunoinformatics is a subset of the field of bioinformatics and computational biology. Specifically, it uses computational, analytical, and mathematical data tied in with computer science processing techniques, to formulate predictions about immunity and vaccine development [14]. In the immunoinformatics-based approach to COVID-19 vaccine development and drug discovery, it is important to note that only the receptor binding domain (RBD) of the SARS-CoV-2 spike protein is in contact with the human ACE2 receptor, making the RBD the major functional region of the virus. Accordingly, the major immunoinformatics-based approaches to COVID-19 antibody development target the RBD of the spike protein, preventing its attachment to the ACE2 receptor [15].
There are two major methodologies of vaccine discovery through immunoinformatics: reverse vaccinology and structural vaccinology [4]. Reverse vaccinology analyzes expressed genomic sequences in order to identify various antigens as potential vaccine targets, as these identified antigens are, ideally, to be synthesized and subsequently targeted by the host immune system. Meanwhile, structural vaccinology uses 3D protein models to engineer immunogenic conformations of antigens in the hopes of eliciting antibody responses against pathogenic attack [16, 17]. Structural vaccinology is not explicitly used in COVID-19 drug discovery, but is incorporated within modern reverse vaccinology techniques [4].
A new study used reverse vaccinology programs as well as novel computation techniques such as the Molecular Mechanics Poisson-Boltzmann Surface Area calculation approach, to design a COVID-19 antibody protein that can provoke a wide array of host immune responses [18]. This immunological approach, in its emphasis on reverse vaccinology, has also been successfully implemented in the design of a multi-epitope subunit vaccine, triggering immunity in both humoral and cell-mediated contexts [19]. Using DNA/PCR visualization software, researchers observed that the multi-epitope vaccine has highly specified, targeted responses to pathogenic invasion via host-mediated immune response [19, 20].
3.2 COVID-19 Docking Analysis
The SARS-CoV-2 docking procedure binds the pathogen to the host’s ACE2 receptors. It is a key point of interest for many researchers who aim to disrupt this binding configuration to prevent COVID-19 infection [21]. Free energy simulators can be used to visualize the stability of various binding configurations of proteins to ligands with a lower binding free energy value indicating a more stable protein-ligand configuration. Using these computational free-energy simulators that bind ligands to the spike protein, potential antibodies can be developed to block or destabilize host-virus interactions [22]. A variety of preliminary studies have been able to identify potential therapeutic compounds from which drug development can progress.
Furthermore, COVID-19 binding can be simulated by docking servers which model how small molecules, peptides, and antibodies bind to potential targets on SARS-CoV-2. In 2020, a team from China created a free meta-server to predict COVID-19 target-ligand interactions to promote drug discovery [23]. This server has been used in a variety of studies. One study used the server to test docking scores of a variety of potential antiviral agents and found that scalarane-based sesterterpenes (a biochemical) showed promise in developing COVID-19 vaccines [24]. Another study, using the same server, identified teicoplanin, an antibiotic, as a potential source of drug design in combating SARS-CoV-2 infection [25].
Several other studies have used docking servers to analyze potential plant-based therapeutic targets; including the compounds of the Boerhavia diffusa, the phytochemicals of the Phyllanthus amarus and Andrographis paniculata, and hesperidin [26-28]. These compounds were initially chosen due to their therapeutic properties and have been previously used to treat a wide array of diseases. Upon further analysis, resulting simulations show that these chemicals can destabilize the ACE2-spike protein complex, thus rendering host immunity [28]. In addition, in silico molecular docking techniques were used in identifying the antiviral drugs Remdesivir and Mycophenolic acid acyl glucuronide as potential candidates to be repurposed towards COVID-19 treatment, due to their preferential binding to the main protease of SARS-CoV-2. This preferential binding can then be used to disrupt the binding of SARS-CoV-2 to human ACE2 receptor cells [29].
3.3 Machine Learning and COVID-19
The field of machine learning is a branch of computer science which trains an algorithm to “learn” through feeding it enough data such that it can make logical predictions about a variety of different sets of conditions [30].
Reverse vaccinology can be combined with machine learning practices to design COVID-19 vaccines. The machine learning tool, known as Vaxign-ML, incorporates biochemical and physicochemical characteristics into its reverse-vaccinology analysis [31]. This platform can then be incorporated with machine learning algorithms, to identify “cocktail” vaccines, consisting of structural and non structural proteins (a protein that is encoded but not part of the viral body), which stimulate an immune response to COVID-19 [32].
Another aspect of machine learning in COVID-19 research involves a more external approach to attacking the problem. Researchers from the Sri Ramaswamy Memorial Institute of Science and Technology were able to train a machine learning algorithm to analyze abnormal chest x-rays and CT scan data in patients exhibiting signs of COVID-19. The study yielded a 93% recall score of CT scan images and 88% precision in analyzing chest x-ray images [33]. Similar machine learning algorithms were used in a different study to analyze abnormal features in the CT scan data of patient lungs. This study yielded an accuracy of 91.94% in diagnosing COVID-19 infection [34].
3.4 MicroRNA (miRNA) and COVID-19 Analysis
MicroRNAs or miRNAs are pieces of RNA which act to regulate gene expression post-transcriptionally [35]. Researchers from Italy and Singapore found that various miRNAs are regulated by the spike protein of SARS-CoV-2 and the human ACE2 receptor in conjunction with the enzyme histone deacetylase (HDAC). Through computational analysis, using in silico methods and the query miRNet analytics platform, these researchers were able to identify that HDAC inhibitors limited interactions between the spike protein and the ACE2 receptor [36]. Further studies confirmed the effectiveness of HDAC inhibitors as a preventative drug to restrict SARS-CoV-2 entrance into the host, using a wide array of laboratory tests and culture analyses [37]. Using similar methodologies, another study constructed its own computational meta-analysis framework to identify how host miRNAs bind to SARS-CoV-2 RNA and suggests the repurposing of anti hepatitis C, RNA based, drugs in the treatment of COVID-19, due to its substantial binding affinity [38].
Current Computational Efforts in COVID-19 Variant Analysis
COVID-19 variants are formed when the virus’ spike protein mutates, making it harder for the established antibodies in vaccinated people to recognize and bind to the pathogen. In some cases, the established antibodies may be able to bind well enough against the variant molecule, while in other cases, a breakthrough infection may occur and the virus is able to override the host’s defense systems [39, 40]. Currently the four major variants of COVID-19 in the United States are: Alpha (B.1.1.7), Beta (B.1.351), Gamma (P.1), Delta (B.1.617.2), and Omicron (B.1.1.529) [41].
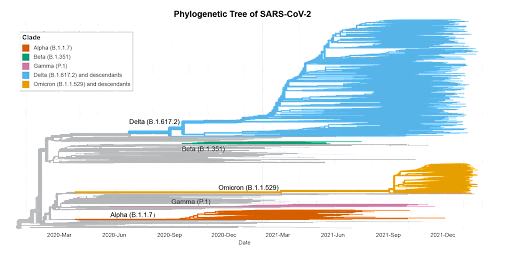
Figure 2: A phylogenetic tree depicting the dominant COVID-19 variants as of December 2021.
By using machine learning algorithms, and comparing genome sequences, researchers were able to obtain an overall picture of the spread of variants throughout all continents [42]. Similarly, other researchers were able to track the global progression of coronavirus variants by aggregating data, concerning the worldwide evolution of COVID-19 nucleotide-substitutions, and building an open source web application known as COVID CG to reflect their findings [43]. Other population-orientated studies investigate the genetic, topological, and evolutionary progression of SARS-CoV-2 in order to understand its emergence on the global scale and how to homogeneously apply vaccines to heterogeneous populations, in the hopes of preventing the spread of COVID-19 and its variants in the future [44-46].
Figure 3: The COVID CG tool, from the end user perspective.
Other studies use computational modeling mechanisms to determine how variants interact with ACE2 receptor cells. One such study modeled a wide array of mutations to the spike protein in order to determine variant transmissibility, which can aid in establishing future safety precautions [47]. Another study was able to model the transmission dynamics of COVID-19 by computationally comparing it with dengue infection (as dengue fever and COVID-19 have similar symptoms in the earlier stages of infection) to obtain alternate insights into COVID-19 disease progression [48].
Conclusion
The field of bioinformatics and computational biology is expansive in its coverage; it can be narrowed down to analyze specific protein-to-protein interactions on the molecular scale or expanded to examine the global progression of disease. With much of the United States reopening its borders, and students returning to in-person classes, the rapid computational analysis of COVID-19 disease progression on both a micro and macro scale is invaluable in ensuring public safety.
As of January 2022, actions to curb the spread of variants have been taken in the form of booster shots and the Pfizer pill. Booster shots reintroduce the same material as the previously mentioned vaccinations to “boost” or reinforce host immunity by increasing the count of memory B and T cells [49]. Additionally, the FDA approved Pfizer COVID-19 Oral Antiviral Treatment, or Paxlovid, consists of nirmatrelvir and ritonavir, with nirmatrelivr acting to prevent viral replication while ritonavir reduces the breakdown of nirmatrelvir. Furthermore, Paxlovid has been proven to be effective against COVID-19 variants in in vitro studies [50-52]. Ultimately, due to the virus’ rapid evolution, most experts have reached a common consensus, that COVID-19 is likely to continually circulate as an endemic, with yearly vaccines needing to be developed and administered, much like the flu [53]. Like the flu, we must continually stay “ahead” of the virus and its variants. Thus, the need for fast, effective computational analysis of the disease and its mutations is essential in mitigating its potentially detrimental effects.
References:
- “WHO Coronavirus (COVID-19) Dashboard.” World Health Organization. Accessed January 4, 2022. https://covid19.who.int/.
- Hwang, Woochang, Winnie Lei, Nicholas M Katritsis, Méabh MacMahon, Kathryn Chapman, and Namshik Han. “Current and Prospective Computational Approaches and Challenges for Developing COVID-19 Vaccines.” Advanced Drug Delivery Reviews 172 (2021): 249–74. https://doi.org/10.1016/j.addr.2021.02.004.
- Wang, May Dongmei. “In The Spotlight: Bioinformatics, Computational Biology and Systems Biology.” IEEE Reviews in Biomedical Engineering 4 (2011): 3–5. https://doi.org/10.1109/rbme.2011.2177935.
- Ishack, Stephanie, and Shari R. Lipner. “Bioinformatics and Immunoinformatics to Support COVID‐19 Vaccine Development.” Journal of Medical Virology 93, no. 9 (2021): 5209–11. https://doi.org/10.1002/jmv.27017.
- Magar, Rishikesh, Prakarsh Yadav, and Amir Barati Farimani. “Potential Neutralizing Antibodies Discovered for Novel Corona Virus Using Machine Learning.” Scientific Reports 11, no. 1 (2021). https://doi.org/10.1038/s41598-021-84637-4.
- Ferrarini, Mariana G., Avantika Lal, Rita Rebollo, Andreas J. Gruber, Andrea Guarracino, Itziar Martinez Gonzalez, Taylor Floyd, et al. “Genome-Wide Bioinformatic Analyses Predict Key Host and Viral Factors in SARS-COV-2 Pathogenesis.” Communications Biology 4, no. 1 (2021). https://doi.org/10.1038/s42003-021-02095-0.
- Martines, Roosecelis B., Jana M. Ritter, Eduard Matkovic, Joy Gary, Brigid C. Bollweg, Hannah Bullock, Cynthia S. Goldsmith, et al. “Pathology and Pathogenesis of SARS-COV-2 Associated with Fatal Coronavirus Disease, United States.” Emerging Infectious Diseases 26, no. 9 (2020): 2005–15. https://doi.org/10.3201/eid2609.202095.
- Parlakpinar, Hakan, and Mehmet Gunata. “SARS‐Cov ‐2 ( COVID ‐19): Cellular and Biochemical Properties and Pharmacological Insights into New Therapeutic Developments.” Cell Biochemistry and Function 39, no. 1 (2020): 10–28. https://doi.org/10.1002/cbf.3591.
- Bettini, Emily, and Michela Locci. “SARS-COV-2 Mrna Vaccines: Immunological Mechanism and Beyond.” Vaccines 9, no. 2 (2021): 147. https://doi.org/10.3390/vaccines9020147.
- Polack, Fernando P., Stephen J. Thomas, Nicholas Kitchin, Judith Absalon, Alejandra Gurtman, Stephen Lockhart, John L. Perez, et al. “Safety and Efficacy of the BNT162B2 Mrna Covid-19 Vaccine.” New England Journal of Medicine 383, no. 27 (2020): 2603–15. https://doi.org/10.1056/nejmoa2034577.
- “CDC Updates and Shortens Recommended Isolation and Quarantine Period for General Population.” Centers for Disease Control and Prevention. Centers for Disease Control and Prevention, December 29, 2021. https://www.cdc.gov/media/releases/2021/s1227-isolation-quarantine-guidance.html.
- “Johnson & Johnson.” Content Lab U.S. Accessed January 4, 2022. https://www.jnj.com/johnson-johnson-announces-real-world-evidence-and-phase-3-data-confirming-strong-and-long-lasting-protection-of-single-shot-covid-19-vaccine-in-the-u-s
- “Johnson & Johnson.” Content Lab U.S. Accessed January 4, 2022. https://www.jnj.com/johnson-johnson-covid-19-vaccine-demonstrates-85-percent-effectiveness-against-hospitalization-in-south-africa-when-omicron-was-dominant.
- Rezaei, Shokouh, Yahya Sefidbakht, and Vuk Uskoković. “Tracking the Pipeline: Immunoinformatics and the COVID-19 Vaccine Design.” Briefings in Bioinformatics 22, no. 6 (2021). https://doi.org/10.1093/bib/bbab241.
- Yang, Jingyun, Wei Wang, Zimin Chen, Shuaiyao Lu, Fanli Yang, Zhenfei Bi, Linlin Bao, et al. “A Vaccine Targeting the RBD of the s Protein of SARS-COV-2 Induces Protective Immunity.” Nature 586, no. 7830 (2020): 572–77. https://doi.org/10.1038/s41586-020-2599-8.
- Graham, Barney S., Morgan S.A. Gilman, and Jason S. McLellan. “Structure-Based Vaccine Antigen Design.” Annual Review of Medicine 70, no. 1 (2019): 91–104. https://doi.org/10.1146/annurev-med-121217-094234.
- Chong, Li C., and Asif M. Khan. “Vaccine Target Discovery.” Encyclopedia of Bioinformatics and Computational Biology, 2019, 241–51. https://doi.org/10.1016/b978-0-12-809633-8.20100-3.
- Enayatkhani, Maryam, Mehdi Hasaniazad, Sobhan Faezi, Hamed Gouklani, Parivash Davoodian, Nahid Ahmadi, Mohammad Ali Einakian, Afsaneh Karmostaji, and Khadijeh Ahmadi. “Reverse Vaccinology Approach to Design a Novel Multi-Epitope Vaccine Candidate against COVID-19: An in Silico Study.” Journal of Biomolecular Structure and Dynamics 39, no. 8 (2020): 2857–72. https://doi.org/10.1080/07391102.2020.1756411.
- Kar, Tamalika, Utkarsh Narsaria, Srijita Basak, Debashrito Deb, Filippo Castiglione, David M. Mueller, and Anurag P. Srivastava. “A Candidate Multi-Epitope Vaccine against SARS-COV-2 – Scientific Reports.” Nature. Nature Publishing Group UK, July 2, 2020. https://doi.org/10.1038/s41598-020-67749-1.
- Tahir ul Qamar, Muhammad, Farah Shahid, Sadia Aslam, Usman Ali Ashfaq, Sidra Aslam, Israr Fatima, Muhammad Mazhar Fareed, Ali Zohaib, and Ling-Ling Chen. “Reverse Vaccinology Assisted Designing of Multiepitope-Based Subunit Vaccine against SARS-COV-2 – Infectious Diseases of Poverty.” BioMed Central. BioMed Central, September 16, 2020. https://doi.org/10.1186/s40249-020-00752-w.
- Hamming, I, W Timens, MLC Bulthuis, AT Lely, GJ Navis, and H van Goor. “Tissue Distribution of ACE2 Protein, the Functional Receptor for SARS Coronavirus. A First Step in Understanding SARS Pathogenesis.” The Journal of Pathology 203, no. 2 (2004): 631–37. https://doi.org/10.1002/path.1570.
- Sixto-López, Yudibeth, José Correa-Basurto, Martiniano Bello, Bruno Landeros-Rivera, Jose Antonio Garzón-Tiznado, and Sarita Montaño. “Structural Insights into SARS-COV-2 Spike Protein and Its Natural Mutants Found in Mexican Population.” Scientific Reports 11, no. 1 (2021). https://doi.org/10.1038/s41598-021-84053-8.
- Kong, Ren, Guangbo Yang, Rui Xue, Ming Liu, Feng Wang, Jianping Hu, Xiaoqiang Guo, and Shan Chang. “Covid-19 Docking Server: A Meta Server for Docking Small Molecules, Peptides and Antibodies against Potential Targets of COVID-19.” OUP Academic. Oxford University Press, July 21, 2020. https://doi.org/10.1093/bioinformatics/btaa645.
- Elhady, Sameh S., Reda F. Abdelhameed, Rania T. Malatani, Abdulrahman M. Alahdal, Hanin A. Bogari, Ahmad J. Almalki, Khadijah A. Mohammad, Safwat A. Ahmed, Amgad I. Khedr, and Khaled M. Darwish. “Molecular Docking and Dynamics Simulation Study of Hyrtios Erectus Isolated Scalarane Sesterterpenes as Potential SARS-COV-2 Dual Target Inhibitors.” Biology 10, no. 5 (2021): 389. https://doi.org/10.3390/biology10050389.
- Azam, Faizul. “Elucidation of Teicoplanin Interactions with Drug Targets Related to Covid-19.” Antibiotics 10, no. 7 (2021): 856. https://doi.org/10.3390/antibiotics10070856.
- Rutwick Surya, U., and N. Praveen. “A Molecular Docking Study of SARS-COV-2 Main Protease against Phytochemicals of Boerhavia Diffusa Linn. for Novel Covid-19 Drug Discovery.” VirusDisease 32, no. 1 (2021): 46–54. https://doi.org/10.1007/s13337-021-00683-6.
- Hiremath, Shridhar, H. D. Kumar, M. Nandan, M. Mantesh, K. S. Shankarappa, V. Venkataravanappa, C. R. Basha, and C. N. Reddy. “In Silico Docking Analysis Revealed the Potential of Phytochemicals Present in Phyllanthus Amarus and Andrographis Paniculata, Used in Ayurveda Medicine in Inhibiting SARS-COV-2.” 3 Biotech 11, no. 2 (2021). https://doi.org/10.1007/s13205-020-02578-7.
- Basu, Anamika, Anasua Sarkar, and Ujjwal Maulik. “Molecular Docking Study of Potential Phytochemicals and Their Effects on the Complex of SARS-cov2 Spike Protein and Human ACE2.” Scientific Reports 10, no. 1 (2020). https://doi.org/10.1038/s41598-020-74715-4.
- Khater, Ibrahim, and Aaya Nassar. “In Silico Molecular Docking Analysis for Repurposing Approved Antiviral Drugs against SARS-COV-2 Main Protease.” Biochemistry and Biophysics Reports 27 (2021): 101032. https://doi.org/10.1016/j.bbrep.2021.101032.
- Badillo, Solveig, Balazs Banfai, Fabian Birzele, Iakov I. Davydov, Lucy Hutchinson, Tony Kam‐Thong, Juliane Siebourg‐Polster, Bernhard Steiert, and Jitao David Zhang. “An Introduction to Machine Learning.” Clinical Pharmacology & Therapeutics 107, no. 4 (2020): 871–85. https://doi.org/10.1002/cpt.1796.
- He, Yongqun, Zuoshuang Xiang, and Harry L. Mobley. “Vaxign: The First Web-Based Vaccine Design Program for Reverse Vaccinology and Applications for Vaccine Development.” Journal of Biomedicine and Biotechnology 2010 (2010): 1–15. https://doi.org/10.1155/2010/297505.
- Ong, Edison, Mei U Wong, Anthony Huffman, and Yongqun He. “Covid-19 Coronavirus Vaccine Design Using Reverse Vaccinology and Machine Learning.” Frontiers in Immunology 11 (2020). https://doi.org/10.3389/fimmu.2020.01581.
- Vinod, Dasari Naga, and S.R.S. Prabaharan. “Data Science and the Role of Artificial Intelligence in Achieving the Fast Diagnosis of Covid-19.” Chaos, Solitons & Fractals 140 (2020): 110182. https://doi.org/10.1016/j.chaos.2020.110182.
- Abbasian Ardakani, Ali, U. Rajendra Acharya, Sina Habibollahi, and Afshin Mohammadi. “COVIDiag: A Clinical CAD System to Diagnose COVID-19 Pneumonia Based on CT Findings.” European Radiology 31, no. 1 (2020): 121–30. https://doi.org/10.1007/s00330-020-07087-y.
- Lu, Thomas X., and Marc E. Rothenberg. “MicroRNA.” Journal of Allergy and Clinical Immunology 141, no. 4 (2018): 1202–7. https://doi.org/10.1016/j.jaci.2017.08.034.
- Teodori, Laura, Piero Sestili, Valeria Madiai, Sofia Coppari, Daniele Fraternale, Marco Bruno Rocchi, Seeram Ramakrishna, and Maria Cristina Albertini. “MicroRNAs Bioinformatics Analyses Identifying HDAC Pathway as a Putative Target for Existing Anti‐Covid‐19 Therapeutics.” Frontiers in Pharmacology 11 (2020). https://doi.org/10.3389/fphar.2020.582003.
- Liu, Ke, Rongfeng Zou, Wenqiang Cui, Meiqing Li, Xueying Wang, Junlin Dong, Hongchun Li, et al. “Clinical HDAC Inhibitors Are Effective Drugs to Prevent the Entry of SARS-COV2.” ACS Pharmacology & Translational Science 3, no. 6 (2020): 1361–70. https://doi.org/10.1021/acsptsci.0c00163.
- Alam, Tanvir, and Leonard Lipovich. “Mircovid-19: Potential Targets of Human Mirnas in SARS-COV-2 for RNA-Based Drug Discovery.” Non-Coding RNA 7, no. 1 (2021): 18. https://doi.org/10.3390/ncrna7010018.
- Hacisuleyman, Ezgi, Caryn Hale, Yuhki Saito, Nathalie E. Blachere, Marissa Bergh, Erin G. Conlon, Dennis J. Schaefer-Babajew, et al. “Vaccine Breakthrough Infections with SARS-COV-2 Variants.” New England Journal of Medicine 384, no. 23 (2021): 2212–18. https://doi.org/10.1056/nejmoa2105000.
- Planas, Delphine, David Veyer, Artem Baidaliuk, Isabelle Staropoli, Florence Guivel-Benhassine, Maaran Michael Rajah, Cyril Planchais, et al. “Reduced Sensitivity of SARS-COV-2 Variant Delta to Antibody Neutralization.” Nature 596, no. 7871 (2021): 276–80. https://doi.org/10.1038/s41586-021-03777-9.
- Aleem, Abdul, Amy K. Slenker, and Abdul Bari Akbar Samad. “Emerging Variants of SARS-COV-2 and Novel Therapeutics against Coronavirus (COVID-19).” National Center for Biotechnology Information. U.S. National Library of Medicine. Accessed January 4, 2022. https://pubmed.ncbi.nlm.nih.gov/34033342/.
- Ekpenyong, Moses Effiong, Mercy Ernest Edoho, Udoinyang Godwin Inyang, Faith-Michael Uzoka, Itemobong Samuel Ekaidem, Anietie Effiong Moses, Martins Ochubiojo Emeje, et al. “A Hybrid Computational Framework for Intelligent Inter-Continent SARS-COV-2 Sub-Strains Characterization and Prediction.” Scientific Reports 11, no. 1 (2021). https://doi.org/10.1038/s41598-021-93757-w.
- Chen, Albert Tian, Kevin Altschuler, Shing Hei Zhan, Yujia Alina Chan, and Benjamin E Deverman. “COVID-19 CG Enables SARS-COV-2 Mutation and Lineage Tracking by Locations and Dates of Interest.” eLife 10 (2021). https://doi.org/10.7554/elife.63409.
- Hahn, Georg, Sanghun Lee, Scott T. Weiss, and Christoph Lange. “Unsupervised Cluster Analysis of Sars‐Cov‐2 Genomes Reflects Its Geographic Progression and Identifies Distinct Genetic Subgroups of SARS‐COV‐2 Virus.” Genetic Epidemiology 45, no. 3 (2021): 316–23. https://doi.org/10.1002/gepi.22373.
- Sarkar, Jnanendra Prasad, Indrajit Saha, Arijit Seal, Debasree Maity, and Ujjwal Maulik. “Topological Analysis for Sequence Variability: Case Study on More than 2K Sars-COV-2 Sequences of COVID-19 Infected 54 Countries in Comparison with SARS-COV-1 and MERS-COV.” Infection, Genetics and Evolution 88 (2021): 104708. https://doi.org/10.1016/j.meegid.2021.104708.
- Tabibzadeh, Alireza, Maryam Esghaei, Saber Soltani, Parastoo Yousefi, Mahsa Taherizadeh, Fahimeh Safarnezhad Tameshkel, Mahsa Golahdooz, et al. “Evolutionary Study of Covid‐19, Severe Acute Respiratory Syndrome Coronavirus 2 (SARS‐COV‐2) as an Emerging Coronavirus: Phylogenetic Analysis and Literature Review.” Veterinary Medicine and Science 7, no. 2 (2020): 559–71. https://doi.org/10.1002/vms3.394.
- Hark Gan, Hin, Alan Twaddle, Benoit Marchand, and Kristin C. Gunsalus. “Structural Modeling of the SARS-COV-2 Spike/Human ACE2 Complex Interface Can Identify High-Affinity Variants Associated with Increased Transmissibility,” 2021. https://doi.org/10.1101/2021.03.22.436454.
- Rehman, Attiq ul, Ram Singh, and Praveen Agarwal. “Modeling, Analysis and Prediction of New Variants of Covid-19 and Dengue Co-Infection on Complex Network.” Chaos, Solitons & Fractals 150 (2021): 111008. https://doi.org/10.1016/j.chaos.2021.111008.
- “Covid-19 Vaccine Booster Shots.” Centers for Disease Control and Prevention. Centers for Disease Control and Prevention. Accessed January 4, 2022. https://www.cdc.gov/coronavirus/2019-ncov/vaccines/booster-shot.html.
- “Pfizer’s Novel Covid-19 Oral Antiviral Treatment Candidate Reduced Risk of Hospitalization or Death by 89% in Interim Analysis of Phase 2/3 Epic-HR Study.” Pfizer. Accessed January 4, 2022. https://www.pfizer.com/news/press-release/press-release-detail/pfizers-novel-covid-19-oral-antiviral-treatment-candidate.
- “Pfizer Receives U.S. FDA Emergency Use Authorization for Novel COVID-19 Oral Antiviral Treatment.” Pfizer. Accessed January 4, 2022. https://www.pfizer.com/news/press-release/press-release-detail/pfizer-receives-us-fda-emergency-use-authorization-novel.
- Commissioner, Office of the. “Coronavirus (COVID-19) Update: FDA Authorizes First Oral Antiviral for Treatment of Covid-19.” U.S. Food and Drug Administration. FDA. Accessed January 4, 2022. https://www.fda.gov/news-events/press-announcements/coronavirus-covid-19-update-fda-authorizes-first-oral-antiviral-treatment-covid-19.
- Torjesen, Ingrid. “Covid-19 Will Become Endemic but with Decreased Potency over Time, Scientists Believe.” BMJ, 2021. https://doi.org/10.1136/bmj.n494.