
Ketamine-Induced Modulation of Glutamatergic Signaling: A Potential Alternative Treatment for Major Depression
Abstract
The widespread prevalence of treatment-resistant depression and inconsistencies in patient responses to traditional serotonergic antidepressants have led to increased interest in glutamatergic antagonists like ketamine as alternative treatments for major depression. Understanding how ketamine acts in the brain to relieve symptoms, as well as its problematic side effects such as dissociation, is a current focus in psychiatric research. Both preclinical and clinical data suggest that ketamine can induce positive changes in behavior and mood by altering glutamate receptor activity and increasing synaptic plasticity, but a specific mechanism has not yet been verified. This review provides an overview of current evidence-based hypotheses for ketamine’s molecular course of action as well as possibilities for further research to reduce dissociative effects. Pharmacological studies in both rodents and humans were selected using the PubMed Advanced Search Builder with keywords ketamine AND antidepressant OR “major depression” and filters for randomized controlled trials within the past five years. Findings from these studies were examined to evaluate the likelihood of ketamine’s interactions with various neural substrates, and how these interactions generate changes in brain activity, mood, and behavior. The data reviewed suggest that ketamine likely acts via multiple neural pathways to increase glutamatergic signaling, and that several pathways can be leveraged to prevent side effects and improve treatment experience for patients.
Keywords: ketamine, antidepressant, NMDA receptor, glutamate, neuroplasticity
Introduction
Major depressive disorder is one of the most prevalent mental health conditions affecting the global population today. The World Health Organization estimates that 5% of adults worldwide suffer from depression, and cites the disorder as one of the leading causes of mental and physical disability [1]. Within the United States, surveys have found that 9.2% of individuals age 12 or older experienced a major depressive episode within the past year [2]. In these studies, depressive episodes are defined by low mood and reduced interest in daily activities that persist for two weeks or longer. Furthermore, over 30% of patients with major depression are treatment-resistant, meaning they do not respond to at least two different antidepressants [3]. Because of this, developing mental health treatments with more consistent outcomes is a significant public health priority. The class of antidepressants most commonly prescribed today, selective serotonin reuptake inhibitors (SSRIs), are thought to relieve depression symptoms by regulating serotonin. However, approximately one third of patients treated with SSRIs will not show a clinically significant increase in mood, and even among those who do respond, symptom relief typically is not experienced until several weeks after starting medication [4, 5]. As a result, many researchers have shifted focus towards alternative paradigms for the biological basis of depression.
One research area with particularly robust supporting evidence is glutamatergic signaling, which is known to play an important role in synaptic plasticity. Synaptic plasticity refers to modifications in the strength of synapses, or connections between neurons, in response to experiences. Glutamate is one of the main chemical messengers that relays excitatory signals across synapses, and changes in expression of receptors for this neurotransmitter are a central mechanism for plasticity in the brain. Findings from a variety of electrophysiology, brain imaging, and post mortem tissue studies implicate dysregulation of glutamatergic signaling as a factor in mood disorder etiology [6]. Pharmacological targeting of glutamate receptors has been found to decrease depression-like behavior in many widely accepted animal models, further corroborating the role of glutamate in major depression. Ketamine, a dissociative anesthetic and derivative of the psychedelic phencyclidine, appears to modify glutamatergic signaling by interacting with one of the major types of glutamate receptors involved in neuroplasticity, N-methyl-D-aspartate (NMDA) receptors or NMDARs. Over the past two decades, ketamine has gained traction among scientists and mental health providers as a potential alternative treatment for depression. In rodent studies, the drug has been found to reduce depressive-type behaviors by strengthening synapses in brain regions relevant to mood [7, 8]. Human trials have also shown that administering subanesthetic ketamine, or a dose lower than what is required for anesthesia, can improve depression symptoms including suicidality and loss of motivation [9]. This relief usually appears within hours of administration, and can last up to twelve days after a single dosage.
However, ketamine’s actions on a molecular level are still not fully understood, and its adverse side effects present a recurrent challenge [7, 8]. Achieving a better understanding of how this drug acts in the brain will be necessary for developing treatments that maximize patient response while minimizing side effects. This review will provide an overview of results from preclinical and clinical trials of ketamine-based therapeutics for major depression. Findings from these studies will shed light on the drug’s clinical relevance, its mechanism of action, and avenues for future pharmaceutical modifications to reduce undesirable side effects.
Ketamine’s Clinical Effects
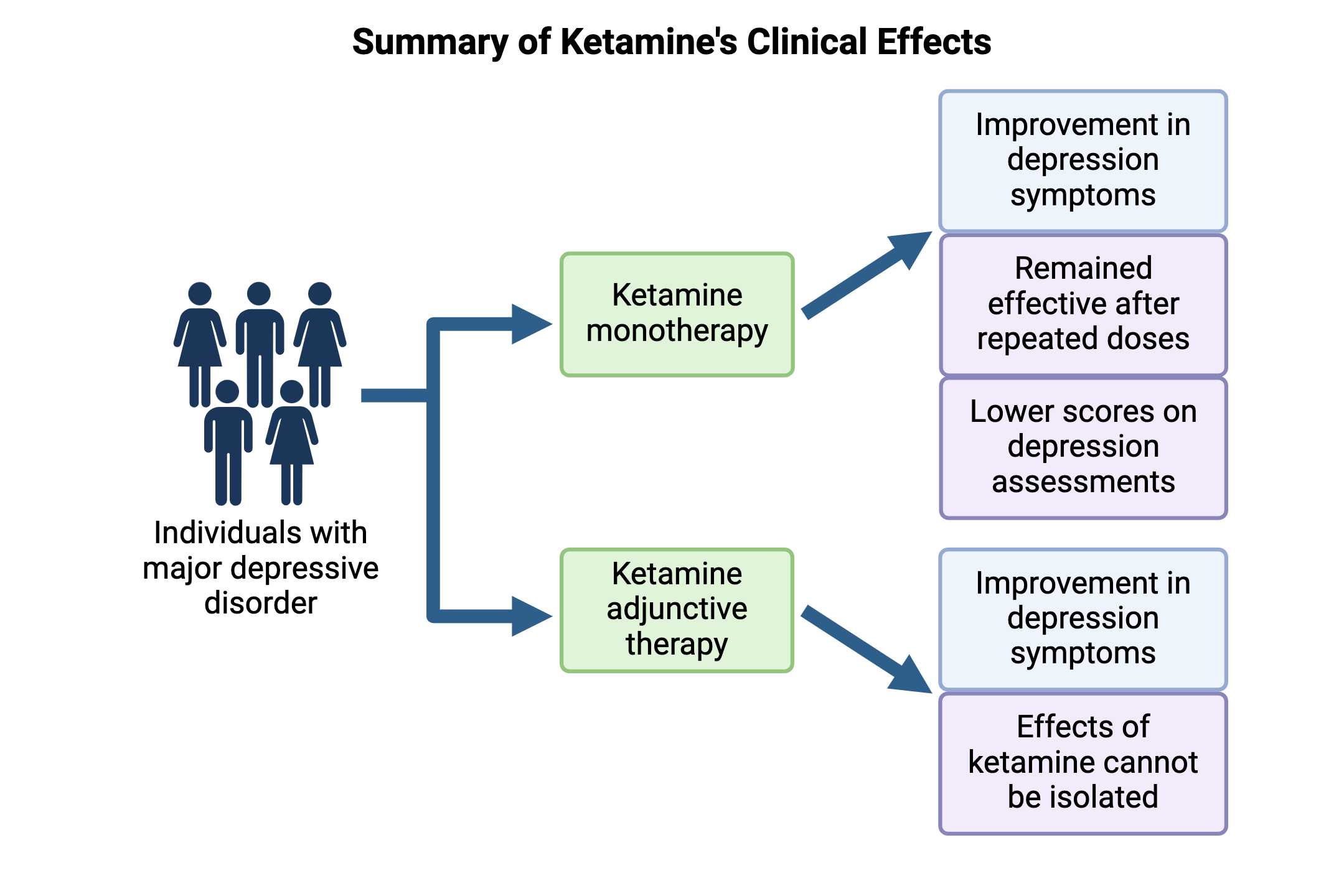
The antidepressant effects of ketamine in humans were first demonstrated by Berman et al in 2000 [10], and many subsequent clinical studies have replicated these results. In 2017, a comparative review of 31 randomized controlled trials found that ketamine generated superior improvement in depression symptoms two weeks after starting treatment when compared with other pharmacotherapies [11]. A retrospective analysis of 537 patients who underwent intravenous ketamine therapy between 2016 and 2020 found that 53.6% of individuals showed a significant response based on decreased scores on the Patient Health Questionnaire, a standardized assessment for monitoring depressive disorders [12]. This questionnaire asks patients to rate the severity of symptoms associated with diagnostic criteria for depression, such as anhedonia, fatigue, altered eating or sleeping patterns, and suicidal ideation. Higher scores indicate more severe depression, while decreases in scores over time indicate remission. In 2022, another systematic review of ketamine in 2665 patients across 79 trials found a similar response rate of 55.3%, and noted that the drug remained effective in repeat doses [13].
Ketamine is also being explored as an adjunctive therapeutic, administered in combination with a second antidepressant. However, this has shown more mixed results than the ketamine monotherapy described above. In 2019, Popova et al [9] found that patients with treatment-resistant depression who were given intranasal ketamine along with a new antidepressant they had not previously tried showed significantly greater symptom improvement compared to patients treated with a placebo nasal spray and a new antidepressant. A clinically meaningful subset of patients also showed improvement within two days of starting the regimen, meaning changes were strong enough to be noticed by patients even if the threshold for statistical significance was not reached. However, studies like these are often unable to isolate the effects of ketamine on mood from the potential effects of the second antidepressant. Therefore, investigating the biological implications of glutamate signaling has been essential in distinguishing between the mechanisms of ketamine and traditional antidepressants.
Ketamine’s Mechanism of Action
Ketamine is a chemical antagonist that binds to NMDARs, preventing glutamate from activating these receptors. This decreased glutamate transmission would theoretically reduce neural activity and synaptic strength, which might be expected to have a negative impact on mood. But paradoxically, ketamine treatment has been found to boost glutamatergic signaling and synaptic efficacy in both preclinical and clinical studies [7, 8]. This section will present several working hypotheses that attempt to reconcile ketamine’s known antagonistic mechanism with these observed effects.
The Direct Inhibition Hypothesis
The direct inhibition hypothesis focuses on the effects of NMDAR blockade on pyramidal neurons (PNs), a class of excitatory neurons found in the cerebral cortex and limbic system. In the absence of ketamine, ambient glutamate levels can activate NMDARs on PNs, triggering an influx of calcium ions into that neuron. These elevated calcium levels induce activation of eukaryotic elongation factor 2 kinase (eEF2K), an enzyme that blocks the activity of the eukaryotic elongation factor 2 (eEF2) protein that plays a central role in peptide synthesis. But when ketamine binds to NMDARs, calcium influx is blocked and eEF2K activity is not enabled, leading to increased eEF2 activity and subsequent synthesis of proteins that contribute to synaptic growth (specifically brain-derived neurotrophic factor, or BDNF). This increase in neuroplasticity through eEF2 and BDNF upregulation is thought to be a major contributor to the drug’s antidepressant properties [7, 8].
The Disinhibition Hypothesis
The disinhibition hypothesis may account for the paradoxical upsurge in glutamatergic signaling induced by ketamine by emphasizing the effects of NMDAR blockade on GABAergic interneurons. In contrast to PNs, GABAergic neurons are inhibitory, meaning their activation reduces the likelihood of other neurons firing. Ketamine appears to preferentially bind to a type
of NMDAR subunit that is highly expressed in corticolimbic GABAergic neurons, and this blockade could reduce inhibition of (i.e. “disinhibit”) activity in the forebrain [7, 8]. Another possible inhibitory mechanism that could explain ketamine’s antidepressant effects is NMDAR antagonism in the lateral habenula (LHb). This brain region inhibits downstream dopaminergic and serotonergic neurons involved in motivation and reward circuitry, and is often overactive in individuals with depression. A 2020 study by Ma et al [14] used in vivo and ex vivo electrophysiological recordings to show that LHb activity is significantly reduced for 24 hours after ketamine injection in mice. Behavioral effects of this ketamine treatment were investigated using the forced swim test (FST) and sucrose preference test (SPT), two of the most widely accepted animal models of depression. The FST uses the amount of time it takes for a rodent to give up trying to escape a cylinder of water as a measure of despair, with shorter mobility times corresponding to depressive-like behavior. Meanwhile, the SPT measures how much preference rodents show for a sucrose solution over water, with reduced preference corresponding to decreased motivation or ability to seek out pleasure. In this study, the mice treated with ketamine showed a marked reduction in behavioral despair and anhedonia in the FST and SPT respectively. These observations suggest a link between ketamine-induced LHb inhibition and relief of depression symptoms.
Convergence on Downstream AMPAR Upregulation
While ketamine’s primary mechanism of action has not been verified, several reviews note the likelihood that both direct inhibition and disinhibition mechanisms play a role in the drug’s antidepressant effects [7, 8]. In both hypotheses described above, increased glutamatergic signaling is thought to upregulate another type of glutamate receptor called α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors, or AMPARs. The downstream effects of higher AMPAR transmission are similar to those of NMDAR blockade in the direct inhibition hypothesis, involving BDNF synthesis and synaptic potentiation [7]. A study by Zhang et al [15] found that hippocampal slices from mice injected with ketamine had increased abundance and phosphorylation of the AMPAR subunit GluA1, which indicates receptor activation, 30 minutes post-injection. These mice also showed reduced behavioral despair in the FST. In addition, studies by Zanos et al and Pałucha-Poniewiera et al [16, 17] both showed that administration of an AMPAR antagonist reversed the behavioral effects of ketamine in mice, suggesting that AMPAR stimulation is necessary for ketamine’s antidepressant mechanism. Overall, it appears highly likely that ketamine acts through multiple complementary pathways to increase neuroplasticity and modulate signaling in mood-relevant brain regions.


Reducing Side Effects
While ketamine therapy was approved by the Food and Drug Administration (FDA) for adults with treatment-resistant depression in 2019 and 2020, concerns over adverse side effects still prevail. Although symptom severity varies greatly across individuals and dosages, typical complications include headaches, dizziness, blurred vision, disorientation, and difficulty speaking [18]. One potential solution to this issue is co-administration of ketamine with another glutamate receptor antagonist, which would allow lower doses of ketamine to be used. In Pałucha-Poniewiera et al’s study [17], combining ketamine with the antagonist LY341495 minimized side effects while retaining antidepressant effects in the FST. However, an important problem is that NMDAR blockade, the mechanism thought to drive ketamine’s antidepressant action, is also responsible for its dissociative side effects. Thus, researchers are currently focused on identifying NMDAR-independent mechanisms whereby the drug might act, which would allow for reduction or elimination of side effects while maintaining an antidepressant response.
Arketamine
One area of focus for NMDAR-independent therapy is using a different chemical form of ketamine. On a molecular level, the drug exists as a mixture of two enantiomers: S-ketamine (esketamine) and R-ketamine (arketamine) [7]. The vast majority of trials to date have used the S-enantiomer, as this molecule has a higher affinity for NMDARs and is therefore more likely to impact glutamatergic signaling. As a result, esketamine is currently the only form of ketamine with FDA approval for clinical use [8].
Because arketamine has a lower NMDAR affinity, higher doses may be required to yield noticeable outcomes, but the reduced binding rate could also lead to less side effects. In 2019, Chang et al [19] compared the antidepressant potential and adverse side effects of arketamine and esketamine in mice. Surprisingly, arketamine had greater potency than esketamine in reversing the effects of chronic social defeat stress based on behavior in the FST, SPT, and social interaction test. Arketamine also led to lower levels of locomotor impairment and acoustic startle reactivity, which are approximations of side effects seen in humans.
In 2021, an unblinded pilot study by Leal et al [20] found that a single dose of arketamine in patients with treatment-resistant depression led to significantly reduced depression scores with negligible side effects. However, subsequent human studies have failed to replicate this outcome: in 2023, a Phase 2a proof-of-concept clinical trial conducted by Atai Life Sciences [21] did not find significant effects of arketamine on depression scores. Later that year, the first double-blinded, placebo-controlled arketamine trial, also led by Leal et al [22], found no significant difference in symptom improvement between arketamine and control groups. Still, reported side effects were far fewer than those in esketamine trials, and these studies have made important contributions to ketamine research by demonstrating that the R-enantiomer is safe for human use.
Hydroxynorketamine
Another avenue of exploration is direct administration of (2R,6R;2S,6S)-hydroxynorketamine (HNK), a metabolite of ketamine. In the body, ketamine is metabolized into HNK within minutes of administration, and while this compound does not inhibit NMDARs, it is still thought to play a
role in ketamine’s antidepressant effects [7, 8]. In a 2019 study by Lumsden et al [23], mice given a HNK injection showed higher hippocampal BDNF levels and reduced behavioral despair in the FST. They also had a decreased latency to eat in the novelty suppressed feeding test (NSF), a behavioral paradigm that uses latency to eat in an unfamiliar environment as a measure of anxiety-like behavior. These findings suggest that HNK administration could bypass ketamine’s dissociative effects while retaining an antidepressant outcome.
Further in vivo studies have allowed researchers to identify a likely mechanism for the antidepressant effects of HNK observed by Lumsden et al. In addition to NMDARs and AMPARs, glutamatergic signaling is regulated by a third category of receptors called group II metabotropic glutamate receptors (mGluRs). One member of this category, mGluR receptor subtype 2 (mGlu2), responds to increased synaptic glutamate levels by blocking further neurotransmitter release via a negative feedback loop. In 2019, Zanos et al [16] found that HNK likely inhibits mGlu2, which would have the overall effect of boosting glutamatergic transmission. In this study, treating mice with a combination of HNK and LY341495, both in doses too low to yield effects on their own, appeared to reduce depressive-type behavior in a synergistic fashion. In another treatment group, mice given both HNK and a glutamate receptor agonist did not show a reduced depressive phenotype, suggesting HNK’s mechanism depends at least in part on mGluR blockade.
Despite this strong preclinical data, a pilot trial by Grunebaum et al [24] of HNK in individuals with suicidal depression found that higher HNK concentrations was inversely correlated with symptom improvement. As with arketamine, future trials will be needed to clarify the antidepressant role of this metabolite in humans.
Conclusion
Ketamine's rapid-onset and long-lasting antidepressant effects in clinical trials have indicated its potential as an alternative therapeutic for treatment-resistant individuals. Rodent studies suggest that ketamine increases glutamatergic signaling and plasticity through a variety of mechanisms including eEF2/BDNF pathway activation, disinhibition of forebrain circuitry, and AMPAR upregulation. While the drug's dissociative effects present a challenge for clinical implementation, the lower NMDAR affinity of arketamine and mGluR antagonist mechanism of HNK are starting points for solutions to this issue.
However, problems with ketamine research persist: Some studies referenced in this review, particularly the arketamine pilot trials [20-22], have questionable statistical power due to highly limited sample sizes. Larger studies will be essential for validating results and making sense of findings that contradict preclinical observations, specifically the antidepressant potency of arketamine and HNK in rodents. Another complication is the difficulty of controlling for factors such as patient medical history and depression severity. For instance, in Popova et al’s study of adjunctive ketamine therapy [9], the novel antidepressant administered differed for each patient depending on what medications they had tried in the past. Ketamine dosage was also individually tailored based on patient symptoms, but this lack of standardization makes it difficult to gather conclusive data on ketamine's pharmacokinetics. Overall, future research will benefit from increased statistical power and more consistent treatment conditions. By continuing to explore the neural pathways impacted by ketamine and investigating NMDAR-independent mechanisms that bypass side effects, researchers can leverage ketamine’s neuroplastic and mood-lifting properties to develop antidepressant treatments that move beyond the traditional serotonin hypothesis of depression.

About the Author: Sinéad Archdeacon
Sinéad is a third-year Neurobiology, Physiology, & Behavior major with a minor in Psychology. Her scientific interests include the biological underpinnings of stress and behavior, genetic tools for studying the brain, and novel developments in mental health treatment. She is currently involved in undergraduate research using single nucleus RNA-seq technology to study the impacts of social stress on oxytocin neurons. After college, she hopes to earn a PhD and pursue a career in neuroscience research. She wrote this review paper as an assignment for her UWP 102B: Writing in the Biological Sciences course, and decided to focus on ketamine therapy for major depressive disorder. Her hope is to provide readers with a better understanding of this important but controversial topic in the mental health field, and spark interest in ongoing medical research that has the potential to improve many people’s lives. In her free time Sinéad enjoys reading, running, spending time with her dogs, and traveling.
Author's Note
I wrote this review paper as an assignment for my UWP 102B: Writing in the Biological Sciences course, which I took in Spring 2024 with Dr. Russ Carpenter. I was asked to write a literature review on a scientific topic of interest, synthesizing previous and current research as well as directions for future investigation. Inspired by my interests in neuroscience and mental health treatment, I decided to write about ketamine therapy for major depressive disorder. My review summarizes findings from rodent and human trials over the past five years, explains the leading hypotheses for how ketamine acts in the brain to alleviate depression symptoms, and explores avenues of ongoing research to minimize the drug’s psychedelic effects in patients. My hope is to provide readers with a better understanding of this important but controversial topic in the mental health field, and spark interest in ongoing neuroscience research that has the potential to improve many people’s lives.
References
World Health Organization. Depressive Disorder (depression). 2023 Mar 31. Accessed 2024 May 1. Available from: https://www.who.int/news-room/fact-sheets/detail/depression.
Goodwin RD, Dierker LC, Wu M, Galea S, Hoven CW, Weinberger AH. 2022. Trends in U.S. depression prevalence from 2015 to 2020: the widening treatment gap. Am J Prev Med [Internet]. 63(5):726–733. doi:10.1016/j.amepre.2022.05.014.
McIntyre RS, Alsuwaidan M, Baune BT, Berk M, Koen Demyttenaere, Goldberg JF, Gorwood P, Ho R, Kasper S, Kennedy SH, et al. 2023. Treatment‐resistant depression: definition, prevalence, detection, management, and investigational interventions. World Psychiatry [Internet]. 22(3):394–412. doi:10.1002/wps.21120.
Cipriani A, Furukawa TA, Salanti G, Chaimani A, Atkinson LZ, Ogawa Y, Leucht S, Ruhe HG, Turner EH, Higgins JPT, et al. 2018. Comparative efficacy and acceptability of 21 antidepressant drugs for the acute treatment of adults with major depressive disorder: a systematic review and network meta-analysis. Lancet [Internet]. 391(10128):1357–1366. doi:10.1016/s0140-6736(17)32802-7.
Alemi F, Min H, Yousefi M, Becker LK, Hane CA, Nori VS, Wojtusiak J. 2021. Effectiveness of common antidepressants: a post market release study. EClinicalMedicine [Internet]. 41. doi:10.1016/j.eclinm.2021.101171.
Price J, Drevets W. 2010. Neurocircuitry of mood disorders. Neuropsychopharmacology [Internet]. 35(1):192-216. doi:10.1038/npp.2009.104.
Zanos P, Gould TD. 2018. Mechanisms of ketamine action as an antidepressant. Mol Psychiatry [Internet]. 23(4):801–811. doi:10.1038/mp.2017.255.
Johnston JN, Bashkim Kadriu, Kraus C, Henter ID, Zarate CA. 2023. Ketamine in neuropsychiatric disorders: an update. Neuropsychopharmacology [Internet]. 49(1):23–40. doi:10.1038/s41386-023-01632-1.
Popova V, Daly EJ, Trivedi M, Cooper K, Lane R, Lim P, Mazzucco C, Hough D, Thase ME, Shelton RC, et al. 2019. Efficacy and safety of flexibly dosed esketamine nasal spray combined with a newly initiated oral antidepressant in treatment-resistant depression: a randomized double-blind active-controlled study. Am J Psychiatry [Internet]. 176(6):428–438. doi:10.1176/appi.ajp.2019.19020172.
Berman RM, Cappiello A, Anand A, Oren DA, Heninger GR, Charney DS, Krystal JH. 2000. Antidepressant effects of ketamine in depressed patients. Biol Psychiatry [Internet]. 47(4):351–354. doi:10.1016/s0006-3223(99)00230-9.
Papadimitropoulou K, Vossen C, Karabis A, Donatti C, Kubitz N. 2017. Comparative efficacy and tolerability of pharmacological and somatic interventions in adult patients with treatment-resistant depression: a systematic review and network meta-analysis. Curr Med Res Opin [Internet]. 33(4):701–711. doi:10.1080/03007995.2016.1277201.
McInnes LA, Qian JJ, Gargeya RS, DeBattista C, Heifets BD. 2022. A retrospective analysis of ketamine intravenous therapy for depression in real-world care settings. J Affect Disord [Internet]. 301:486–495. doi:10.1016/j.jad.2021.12.097.
Alnefeesi Y, Chen-Li D, Krane E, Jawad MY, Rodrigues NB, Ceban F, Di Vincenzo JD, Meshkat S, Ho RCM, Gill H, et al. 2022. Real-world effectiveness of ketamine in treatment-resistant depression: a systematic review & meta-analysis. J Psychiatr Res [Internet]. 151:693–709. doi:10.1016/j.jpsychires.2022.04.037.
Ma S, Chen M, Jiang Y, Xiang X, Wang S, Wu Z, Li S, Cui Y, Wang J, Zhu Y, et al. 2023. Sustained antidepressant effect of ketamine through NMDAR trapping in the LHb. Nature [Internet]. 622:802-809. doi:10.1038/s41586-023-06624-1.
Zhang K, Xu T, Yuan Z, Wei Z, Vítor Nagai Yamaki, Huang M, Huganir RL, Cai X. 2016. Essential roles of AMPA receptor GluA1 phosphorylation and presynaptic HCN channels in fast-acting antidepressant responses of ketamine. Sci Signal [Internet]. 9(458). doi:10.1126/scisignal.aai7884.
Zanos P, Highland JN, Stewart BW, Georgiou P, Jenne CE, Lovett J, Morris PJ, Thomas CJ, Moaddel R, Zarate CA, et al. 2019. (2R,6R)-hydroxynorketamine exerts mGlu2 receptor-dependent antidepressant actions. Proc Natl Acad Sci USA [Internet]. :201819540. doi:10.1073/pnas.1819540116.
Pałucha-Poniewiera A, Podkowa K, Pilc A. 2019. Role of AMPA receptor stimulation and TrkB signaling in the antidepressant-like effect of ketamine co-administered with a group II mGlu receptor antagonist, LY341495, in the forced swim test in rats. Behav Pharmacol [Internet]. 30(6):471–477. doi:10.1097/FBP.0000000000000471.
Acevedo-Diaz E, Cavanaugh G, Greenstein D, Kraus C, Kadriu B, Zarate C, Park L. 2020. Cognitive reactivity predicts depressive symptoms in everyday life: an experience sampling study. J Affect Disord [Internet]. 266: 545–552. doi:10.1016/j.jad.2019.11.028.
Chang L, Zhang K, Pu Y, Qu Y, Wang S, Xiong Z, Ren Q, Dong C, Fujita Y, Hashimoto K. 2019. Comparison of antidepressant and side effects in mice after intranasal administration of (R,S)-ketamine, (R)-ketamine, and (S)-ketamine. Pharmacol Biochem Behav [Internet]. 181:53–59. doi:10.1016/j.pbb.2019.04.008.
Leal GC, Bandeira ID, Correia-Melo FS, Telles M, Mello RP, Vieira F, Lima CS, Jesus-Nunes AP, Guerreiro-Costa LNF, Marback RF, et al. 2020. Intravenous arketamine for treatment-resistant depression: open-label pilot study. Eur Arch Psychiatry Clin Neurosci [Internet]. 271:577-582. doi:10.1007/s00406-020-01110-5.
GlobeNewswire. Atai Life Sciences Announces Results from Phase 2a Trial of PCN-101 (R-ketamine) for Treatment-Resistant Depression. 2023 Jan 6. Accessed 2024 Jun 2. Available from: https://www.globenewswire.com/news-release/2023/01/06/2584334/0/en/atai-Life-Sciences-Announces-Results-from-Phase-2a-Trial-of-PCN-101-R-ketamine-for-Treatment-Resi stant-Depression.html.
Leal GC, Souza-Marques B, Mello RP, Bandeira ID, Caliman-Fontes AT, Carneiro BA, Faria-Guimarães D, Guerreiro-Costa LNF, Jesus-Nunes AP, Silva SS, et al. 2023. Arketamine as adjunctive therapy for treatment-resistant depression: a placebo-controlled pilot study. J Affect Disord [Internet]. 330:7–15. doi:10.1016/j.jad.2023.02.151.
Lumsden EW, Troppoli TA, Myers SJ, Zanos P, Aracava Y, Kehr J, Lovett J, Kim S, Wang F-H, Schmidt S, et al. 2019. Antidepressant-relevant concentrations of the ketamine metabolite (2R,6R)-hydroxynorketamine do not block NMDA receptor function. Proc Natl Acad Sci USA [Internet]. 116(11):5160–5169. doi:10.1073/pnas.1816071116.
Grunebaum MF, Galfalvy HC, Choo T-H, Parris MS, Burke AK, Suckow RF, Cooper TB, Mann JJ. 2019. Ketamine metabolite pilot study in a suicidal depression trial. J Psychiatr Res [Internet]. 117:129–134. doi:10.1016/j.jpsychires.2019.08.005.